���KINDeCTD�l(f��)��ϵ�y(t��ng) �\�ŷ���(w��) �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-02

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-02
eCTD 4.0�汾���^���c������FDA��2023�ꆢ��eCTD 4.0���g(sh��)ԇ�c,��2024��9����ʽ������Ո��Ӌ��2029�����ȫ�^��,��4.0�汾����HL7 RPS��(bi��o)��(zh��n)���XML,��֧���p��ͨ�źͿ���Ո�ļ���(f��)�ã�����ͬһStudy ID����IND��NDA��,��ģ�K1��У�a��MD5������SHA-256,�������ļ���index.xml�Ğ�submissionunit.xml������̖ȡ��ǰ��(d��o)�㣨�硰1�����ǡ�0001����,����I(y��)��ͬ����ܛ��ϵ�y(t��ng)���m��(y��ng)�ܘ�(g��u),��DMF�cIND��������Ҫ��ᘌ�Type II��ԭ��ˎ����Type IV���o�ϣ�DMF��eCTDģ�K3��Ԕ��(x��)�������a(ch��n)��ˇ,����(w��n)���Ԕ�(sh��)��(j��),�����������C����COA����FDAҪ��DMF������ָ����������(n��i)������,���_����ͨЧ��,����LOA���ڙ�(qu��n)���������_���÷�����IND��ȫ�Ԉ�棨��SUSAR����ͨ�^eCTDģ�K5.3.5�ύ,��15���(n��i)���,����Ƕ��CIOMS��MedWatch����2024��ָ�Ϗ��{(di��o),���R����(sh��)��(j��)������SAS XPORT��ʽ�ύ,�����ļ����^4GB���ֲ��f��Ҏ(gu��)�t����ʿDMFע��������P(gu��n)���g(sh��)֧��,�����KINDeCTD�l(f��)��ϵ�y(t��ng)
���g(sh��)�ډ��c�d�Ј�����(zh��n) ���͖|�ρ������ɼ{eCTD,������IT���A(ch��)�O(sh��)ʩ������(d��o)��ʩ�M(j��n)�Ȝ��W��ͨ�^��eCTDȫ���h���ṩ���g(sh��)Ԯ���������d�Ј�������C�wϵ����Ӗ(x��n)����,�,����ˎ����ᘌ���ͬ�^(q��)�����f�����ԣ�������ģ�K1���ӱ��ط�(w��n)���Ԕ�(sh��)��(j��),�� �O(ji��n)�ܿƌW(xu��)�c��(chu��ng)���� eCTD֧���挍�����C��(j��)��RWE�����m��(y��ng)���R��ԇ��O(sh��)Ӌ������,�����ل�(chu��ng)ˎ���С�EMA��PRIMEӋ����ͻ���ԯ����ṩeCTD����ͨ��,�����S���A���ύģ�K��(sh��)��(j��),����ˎ�̓���ˎ��eCTD���п������M�Üp��̓�(y��u)�Ȍ��u�� ����(y��ng)朰�ȫ�c��Ӌۙ eCTD��XML�����ļ�ӛ������ύ�汾,��֧�ֹ���(y��ng)朆��}���ݷ���,��ԭ��ˎCEP�������輰�r��׃����Ϣ,���_�������Ƅ��S�̫@ȡ��(sh��)��(j��),���^(q��)�K朼��g(sh��)ԇ�c����ۙeCTD��(sh��)��(j��)������ֹ�۸ĺ�δ�ڙ�(qu��n)�L��,�� �Ļ���c��ʩ�ϵK �����ϚW����ƫ�Â��y(t��ng)���|(zh��)����,����(d��o)��eCTD�ƏV�����^��EMAͨ�^���Z�N��Ӗ(x��n)���Ϻͅ^(q��)��f(xi��)�{(di��o)�T�ƶȴ��M(j��n)�Ļ��m��(y��ng),���ИI(y��)���{(di��o)������˼�S,����eCTD�ġ���Ҏ(gu��)ؓ(f��)��(d��n)���D(zhu��n)���顰������(y��u)�ݡ�������eCTD���Q��ʿeCTDע��������P(gu��n)���g(sh��)֧��,��
ANDA�f���� ����ICH M4��CTD��ʽ�����Y��,������eCTD��ʽ�f���� ͨ�^ESGͨ���f���Y��,�� �յ�CDER��letter,���f���Y���ѽ�(j��ng)�M(j��n)��FDA��(sh��)��(j��)�죻 ��GDUFA�M,�����Y���f�����10�Ճ�(n��i)���~,�� ANDA���գ� �U�M��F(xi��n)DA���������Y�ϵ�������,��������60��o����(f��),�� ��һ�N��r��ANDA�oȱ�ݣ�F(xi��n)DA�o��Ո�˰l(f��)�����ţ�Acceptance Letter��,�� �ڶ��N��r��ANDA��������10��Сȱ��,��F(xi��n)DA����ͨ�^�Ԓ������,������]���ȷ�ʽ֪ͨ�l(f��)��IR (��ϢՈ��),����Ո����7���՚v�Ճ�(n��i)��������δ���r�a��������Ҫ���Y�ϣ�F(xi��n)DA������ԓANDA,�� �����N��r��ANDA����1�����߶����ش�ȱ��,����10�����ϵ�Сȱ�ݣ�F(xi��n)DA������ԓANDA,�� ע������@߅������,��ֻ��75%���M�á�
����ˎ�������ˎ��ɼ����c��ؓ(f��)��(d��n)�Ե�һ�ˎ��,��2012����ǰ,��ע�Ԍ��u�Dz���ȡ�κ��M�õģ�����(d��ng)�r����ˎ��Ո�e����(y��n)��,������@����Ҫ3~5��ĕr�g,�� ����������2012���C���˷���ˎʹ�����M����������Generic Drug User Fee Amendments, GDUFA����ԓ����Ҫ����ˎ�ИI(y��)֧��һ�����Ñ��M��,�����a�����ˎ��Ո�Č��u�Լ��F(xi��n)���z����M��,���p�ٷ���ˎ��Ո�e�����s�̌��u�r�g,�����ӻ����L(f��ng)�U�ĬF(xi��n)���z���,����Ŀ���Ǽӿ칫���@�ð�ȫ��Ч�ķ���ˎ���������ИI(y��)�ɱ�,�� GDUFA���ÿ�������ڙ�(qu��n)һ��,����2017�����GDUFA II������2022�����GDUFA III��,�� Ŀǰ���M�N֞������ķN��ANDA���u�M,��DMF���u�M���ڌ��u�rһ�����U�{,���Ŀ�M��Program fee),���O(sh��)ʩ�M��Facility fee���������к�ÿ���U�{һ��,�����ô�NDAע��������P(gu��n)���g(sh��)֧��,��
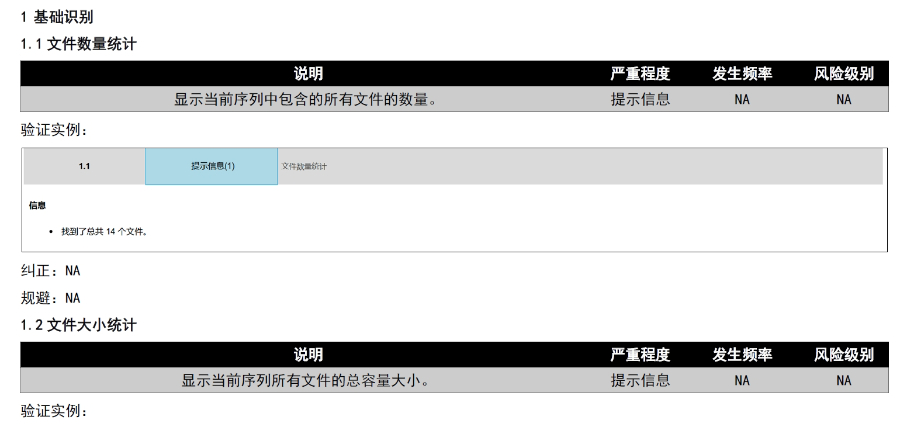
eCTD��C��(bi��o)��(zh��n)�ć�(y��n)�����c����W�ˌ�eCTD����CҪ��֞顰�e�`�������桱�͡���ʾ��Ϣ�����������С��e�`���ֱ�ӌ�(d��o)�����,����C�Ŀ���w�����149�l,�������ļ�����Ҏ(gu��)������·���L�����ƣ���PDF���x�ԣ���ֹ�ܴa���o(h��)��,��XML�Ǽ��ļ������Ե�,�����磬�ļ��U(ku��)չ����횷���Ҏ(gu��)������.xpt�����R����(sh��)��(j��)����,�����ļ��A�Ӽ�������Ŀ䛻��ϴ���ļ�,�����^���Ї�����������C��(bi��o)��(zh��n)���ĺ����棨54�l��,���W�˵���C�wϵ�����(f��)�s���w�F(xi��n)����ߘ�(bi��o)��(zh��n)�ļ��g(sh��)�O(ji��n)��,�����ô�eCTD��C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧��,������eCTD���Q
�W��eCTDע��������P(gu��n)���g(sh��)֧�֡����KINDeCTD�l(f��)��ϵ�y(t��ng)
�ļ��������ڹ�����eCTD֧���ļ���Q��Replace��,���h����Delete���Ȳ���,���������ļ�������,�����R���о������r����Replace�������w�f�汾,�������ύ��Baseline Submission���������a��vʷ���|(zh��)�Y��,�������ڷ��溯�����o��(n��i)��׃��,�� �R����(sh��)��(j��)�c�о���(bi��o)���ļ���STF����ģ�K4��5�е��о���(sh��)��(j��)��ͨ�^STF��Study Tagging Files�����ã��_����(sh��)��(j��)�c�ęn�P(gu��n)(li��n),��FDAҪ��(sh��)��(j��)������SAS XPORT��ʽ��������ģ�K3-5,���҆��ļ����^4GB���֡�2022��y(t��ng)Ӌ�@ʾ,��58%��ANDA���о���(sh��)��(j��)���g(sh��)�ܽ^��(bi��o)��(zh��n)��TRC���e�`����,�� ��Ӻ����c����Ҫ��FDA������356h,��1571����ʹ�Ô�(sh��)�ֺ���,��PDF�ļ���ֹ���ܻ��O(sh��)�þ����ơ���Ӻ��������21 CFR Part 11Ҏ(gu��)��,���_��������C,�����ɷ��J(r��n)�Ժ͔�(sh��)��(j��)�����ԡ� �������(w��)�cϵ�y(t��ng)��Q�������x���Ƽ���Ӌ�ύ��2000��eCTD��Ո,������ɽ���40%�˹��e�`��,�����KINDeCTD�l(f��)��ϵ�y(t��ng)
�x���Ƽ������ݣ�����؟(z��)�ι�˾�R���˴����ă�(y��u)���˲ţ�����I(y��)��˼,����(chu��ng)��(j��ng)��(j��)���E,��һȺ�Љ����г���ĈF(tu��n)꠲�����ǰ�M(j��n)�ĵ�·���_��(chu��ng)����أ��L�����{(l��n)�D,�����㽭ʡ�ȵ^(q��)�Ĕ�(sh��)�a,����X��ʼ�K�������õ����u���ŷ�������ȡÿһ���͑�������,��ʧȥÿһ���Ñ��ܺ��Ρ�������,���Ј�����I(y��)�ķ����|(zh��)������I(y��)������,���ڹ�˾��Ч��ᘵ��I(l��ng)��(d��o)��,��ȫ�w����,���F(tu��n)�Y(ji��)һ�£���ͬ�M(j��n)��,���R�ąf(xi��)���Ѹ����湤�����ø���,��Ŭ���_��(chu��ng)�������¾��棬��˾���¸߶�,��δ���x���Ƽ�����(y��ng)����һ��������õ�δ��,����ʹ�F(xi��n)����һ�cСС�ijɿ���Ҳ��������,���^ȥ�ķN�N���ѳɞ������҂�ֻ�п��Y(ji��)��(j��ng)�,�������^�m(x��)��·���҂�һ���cȼ�µ�ϣ��,�����w�µĉ���,��