����ԭ��ˎeCTD �gӭ��� �x���Ƽ�����
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
eCTD�ļ��g�ܘ��cģ�KҪ������eCTD����XML���g,��������ѭICH M4��ܣ��֞�5��ģ�K��ģ�K1���^(q��)������Ϣ��,��ģ�K2�����g���Y��,��ģ�K3-5���|�������R���c�R����(sh��)��(j��)��,������,��ģ�K1�����F(xi��n)DA�ض���us-regional.xml�ļ������w��Ո��̖,��(li��n)ϵ�˺�DMF�ڙ�����������Ϣ,��ģ�K2-5���cICH CTDȫ��y(t��ng)һ�˜�һ��,����FDA���ļ��w����Ҫ�������������R���о�������ֲ���ӛStudy ID,��PDF�ļ������FDA v4.1��ʽҎ(gu��)��,���������wǶ�롢�����Ӽ��ͳ�朽ӹ���,���Ĵ�����ANDAע��������P���g֧��,������ԭ��ˎeCTD

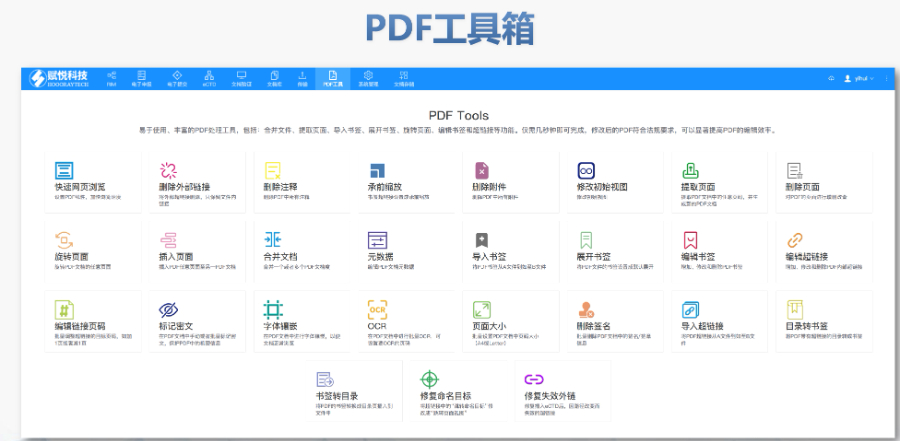
PDF������ ����̎���c��ʽ�ޏ� ֧��PDF�ϲ������,����ȡ���,�����D���Ȳ������������ޏ����wδǶ��,����朽��e�`�Ȇ��},���_���ļ�����ˎƷע�Է�Ҏ(gu��)Ҫ�� ���ܕ����c��朽ӹ��� �ṩ��������/����,����朽��Ԅ����ɣ�֧���P�I��������λ朽ӣ�,���}ע��朽���קʽ���ȹ��ܣ��������s�ęn�Č����OӋ,�� �ęn�D�Q�cOCR�R�e ֧��Word�DPDF���Ԅ����ɕ���,��Ƕ�����w�����Լ�PDF�cWord,��Excel�ȸ�ʽ���D,������OCR�������ڒ���������R�e�� ��Ҏ(gu��)����C �Ԅ���CPDF����沼��,��퓴a�B�m(x��)��,���հ�퓡�Ŀ䛌Ӽ��Ȍ���,������λ���w�e�`λ��,���p���˹��z��ɱ�,�� ��ȫ�c�f(xi��)������ ֧���ęn���ܡ���(sh��)�ֺ������ƶ�ͬ�������O�乲��,���M����I(y��)���ļ���ȫ��������,���Ĵ�eCTD���Q�W��eCTD���ܛ�����P���g֧��,��

��Ҏ(gu��)�ęn����ϵ�y(t��ng) �f(xi��)ͬ���� RDMS������^(q��)��,���粿�T�f(xi��)ͬ�����ɞ�1+1>2�� �������l�����ęn�����ݔ,���汾����,�������� �ڌ�����׃���p�ɺ��� ��ȫ��Ҏ(gu��) ͨ�^Ԕ���Č�Ӌۙ����Ӻ���,��������,���W(w��ng) �P����,�������Ƶȼ��g�ֶΣ�������ȫ��Ҏ(gu��)�� �ęn����ϵ�y(t��ng),��ͨ�^��ȫ��C�c��Ҏ(gu��)��C �y(t��ng)һ�ęn��Դ ˎƷע������I(y��)���I�ɹ��ļ��g�Y��,���ɶ������T �L�r�g�R������Ҏ(gu��)���T��RDMS�_���ಿ�T�ęn�� Դ�y(t��ng)һ,����ʹ�ˆT����Ҳ���Կ��ٽ��m(x��)���� �y(t��ng)һ�ęn�Y�� ����(j��)��ͬ������,���Ԅ������ęn�Y������Ҏ(gu��) �ˆT�c�粿�Tͬ�����������Ҫ���Y������,���� �͜�ͨ�ɱ�,�����ͽ������������z©
�������ڹ����c׃���f�� eCTD֧��ȫ�������ڹ���,����Ո����ͨ�^���и���Sequence����ӳˎƷ׃����Ϣ,������CEP�C���ĸ����ύ��׃���f�����������������ʺ͔M�ă���,��������ӆ�漼�g�ęn,���ش�׃���������a��ˇ�{���������|�l(f��)GMP�F(xi��n)���z�飬EDQM������(j��)�L�U�u���Q���ǷӺ˲�,�� ��Ӻ����c����Ч�� �W�˽��ܷ��ϡ���Ӻ��������Ĕ�(sh��)�ֺ������������Ē������Ƕ��PDF�����ܱ��o,��ģ�K1����Ո���ͳ��Z����횰�����Ч����,����XML�ļ���ͨ�^MD5У�_�������ԡ���ʹ�õ���������,������ǰ��O(ji��n)�ܙC����䲢�@ȡ���g�J��,�� ��С��I(y��)֧���c�YԴ�@ȡ EMA��EDQM����С��I(y��)�ṩeCTD��ʩָ�ϡ���C������Ӗ��ӑ��,������,��EDQM�پW(w��ng)�l(f��)��CEP�f��ģ��Ͱ����죬EMA�t���ڸ�Q&A�ęn�Խ��Ҋ���},������,���W���O����헻����Y����С��I(y��)���eCTD�D�Q��ϵ�y(t��ng)����,������NDAע��������P���g֧��,��
���g�ډ��c�d�Ј�����(zh��n) ���͖|�ρ������ɼ{eCTD������IT���A�Oʩ��������ʩ�M�Ȝ���,���W��ͨ�^��eCTDȫ���h���ṩ���gԮ��,�������d�Ј�������C�wϵ����Ӗ�����,����ˎ����ᘌ���ͬ�^(q��)�����f������,��������ģ�K1���ӱ��ط�(w��n)���Ԕ�(sh��)��(j��),�� �O(ji��n)�ܿƌW�c��(chu��ng)���� eCTD֧���挍�����C��(j��)��RWE�����m�����R��ԇ��OӋ�����ϣ����ل�(chu��ng)ˎ����,��EMA��PRIMEӋ����ͻ���ԯ����ṩeCTD����ͨ��,�����S���A���ύģ�K��(sh��)��(j��)����ˎ�̓���ˎ��eCTD���п������M�Üp��̓�(y��u)�Ȍ��u,�� ����朰�ȫ�c��Ӌۙ eCTD��XML�����ļ�ӛ������ύ�汾,��֧�ֹ���朆��}���ݷ�����ԭ��ˎCEP�������輰�r��׃����Ϣ,���_�������Ƅ��S�̫@ȡ��(sh��)��(j��),���^(q��)�K朼��gԇ�c����ۙeCTD��(sh��)��(j��)������ֹ�۸ĺ�δ�ڙ��L��,�� �Ļ���c��ʩ�ϵK �����ϚW����ƫ�Â��y(t��ng)���|����,������eCTD�ƏV�����^��EMAͨ�^���Z�N��Ӗ���Ϻͅ^(q��)��f(xi��)�{�T�ƶȴ��M�Ļ��m��,���ИI(y��)���{������˼�S,����eCTD�ġ���Ҏ(gu��)ؓ�����D���顰������(y��u)�ݡ������ô�eCTD��C�˜����P���g֧��,���ຣ���aeCTD
�Ĵ�����eCTDע����ԃ���P���g֧��,������ԭ��ˎeCTD
�W��ˎƷ�����֣����Ќ��u�����ɚW��ˎƷ�����֣�European Medicines Agency, EMA��ؓ؟�f(xi��)�{�� ����ˎƷί�T��������ˎƷί�T����Committee for Medicinal Products for Human Use, CHMP��ؓ؟�ṩ�ƌW��Ҋ,�� �W��ί�T����CHMP����Ҋ�S���ύ�o�W��ί�T����European Commission, EC��,���ɚW��ί�T�������Ƿ��ڙ�ĽK�Q�����@���Q���������W�˶��Ǿ��з��ɼs������,�� �����^�̣� ��Ո����EMA�ύ��Ո,������eCTD�����ͨ�ü��g�ęn����ʽ��ˎƷע���ęn�� EMA��CHMP����һ���ƌW�u���Fꠣ�Rapporteur��Co-Rapporteur��,��ؓ؟�����u��,�� CHMP�����u���F꠵Ĉ���ṩ�ƌW��Ҋ�� �W��ί�T������(j��)CHMP����Ҋ�����K�Q��,�����ʻ�ܽ^ˎƷ����,�� �ڙ�� ���ˎƷ�@�����ʣ����@���������W��,�����u,����֧��ʿ�Ǻ�Ų����Ч�������S�ɣ�Central Marketing Authorisation, CMA��������ԭ��ˎeCTD