ɽ�|������ƷeCTD ���C���A �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-03

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-03
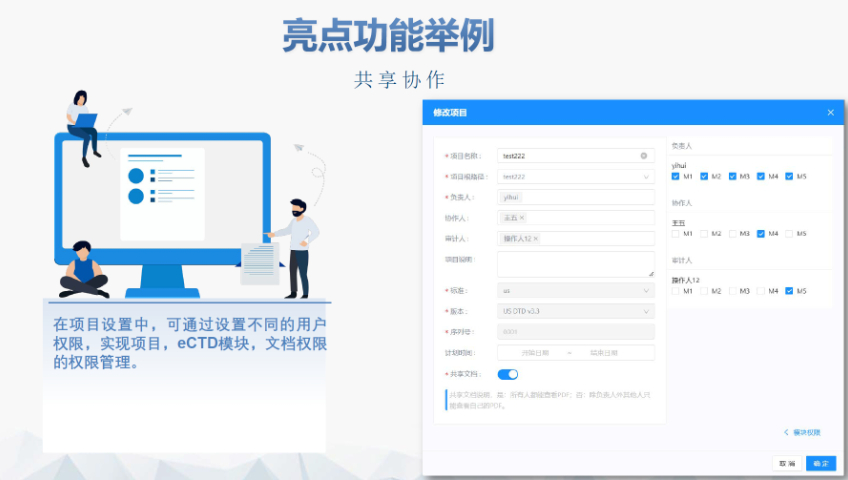
�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ��,������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)�,�����J(r��n)����MRP����,�������ɆT����RMS�����u������豻�����ɆT���J(r��n)��,�������F(xi��n)��������CMDh�f(xi��)�{(di��o)���ύEMA�ٲá��@�N���Ӽ����u�C(j��)��Ҫ����Ո�����ļ���(zh��n)���A�μ����]�^(q��)�������,��������m(x��)�������`,�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y(ji��)��(g��u)��֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е�����,��������(qi��ng)�������ڹ�������,���W��Ӌ��ͨ�^2024��ԇ�c���^����4.0�����ƽ���ļ��M����ʽ�����p���؏�(f��)�ύ���������uЧ��,��Ȼ��,����ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m��(y��ng)�Ԇ��}����NDAע��������P(gu��n)���g(sh��)֧��,��ɽ�|������ƷeCTD

ANDA�f���� ����ICH M4��CTD��ʽ�����Y��,������eCTD��ʽ�f���� ͨ�^ESGͨ���f���Y��,�� �յ�CDER��letter,���f���Y���ѽ�(j��ng)�M(j��n)��FDA��(sh��)��(j��)�죻 ��GDUFA�M,�����Y���f�����10�Ճ�(n��i)���~,�� ANDA���գ� �U�M��F(xi��n)DA���������Y�ϵ�������,��������60��o����(f��),�� ��һ�N��r��ANDA�oȱ�ݣ�F(xi��n)DA�o��Ո�˰l(f��)�����ţ�Acceptance Letter��,�� �ڶ��N��r��ANDA��������10��Сȱ��,��F(xi��n)DA����ͨ�^�Ԓ������,������]���ȷ�ʽ֪ͨ�l(f��)��IR (��ϢՈ��),����Ո����7���՚v�Ճ�(n��i)����,����δ���r�a(b��)��������Ҫ���Y�ϣ�F(xi��n)DA������ԓANDA,�� �����N��r��ANDA����1�����߶����ش�ȱ��,����10�����ϵ�Сȱ�ݣ�F(xi��n)DA������ԓANDA,�� ע������@߅������,��ֻ��75%���M��,���㽭�x���Ƽ�eCTD��ʲô��ʿeCTD��C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧��,��
�W��eCTD��ʩ��(j��ng)��S�����Ї��ɽ��b�Լ����M(j��n)��,���Ї����ܕ���(j��ng)�v����I(y��)��ԸeCTD�ύ����(qi��ng)��eCTD�ύ���^��,���Ҍ��o�SICH������������CMC�Y����������,��ȫ������eCTD 4.0�^��,���Ї�Ҳ�����⣬���S�ձ�,���W��,�������ȏ�(qi��ng)�ƌ�ʩ�������M(j��n)�� �Ї���������ȫ�����C(j��)��,��eCTD��ʩ�������Ї���I(y��)��������,�����g(sh��)�M(j��n)��������eCTD��ʩ����I(y��)�������P(gu��n)ע���g(sh��)�ӑB(t��i),���{(di��o)����(zh��n)��,���S������(n��i)�͘I(y��)�����������I(y��)�����������ӣ��˽�eCTD�ȇ��H��(bi��o)��(zh��n)���ɞ��I(y��)�l(f��)չ����Ҫ������,�� �Ї����M(j��n)eCTD���挦��ɫ���},�������к���Ո�Y��ƥ�䣬����I(y��)�c�O(ji��n)�ܙC(j��)��(g��u)��ͬ��Q,�,�����Ҫ�����P(gu��n)�I���Ї����ƶ��m�χ����Ҫ��,���ڴ�δ���(zh��)��ָ�ϼȾ���ɫ���c���H��܉,����eCTD��ʩ�ṩ֧�֡�
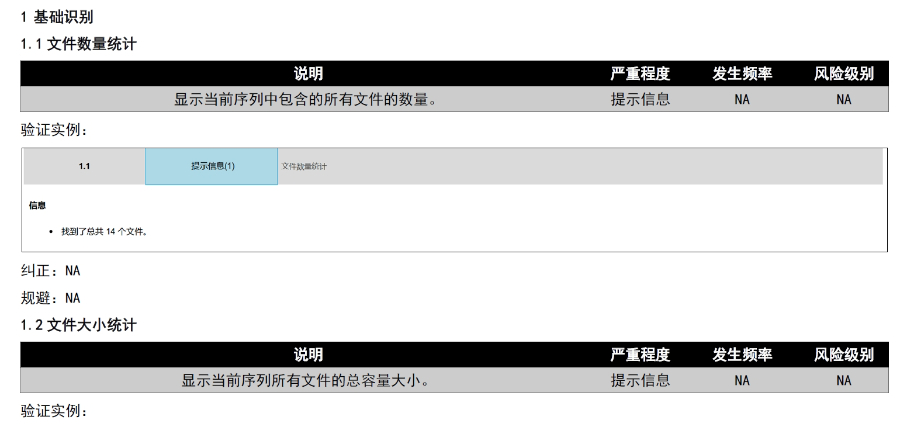
�������u�����ceCTD�f��;�����m�䣺�W��ˎƷ���u����������У�CP��,����ɢ��DCP��,�����J(r��n)��MRP���͇��ҳ���NP����eCTD���m�䲻ͬ������f��Ҫ��,�����磺 ���Ќ��u����CP����ͨ�^EMA��eSubmission Gateway�ύ,�����u�r�s240��������,��eCTD�����������ģ�K1-5�����Z�Ԙ�(bi��o)���ļ��� ��ɢ���u����DCP������ͨ�^CESP���W�˹�ͬ�ύ�T�����f��,�������ɆT����RMS������(d��o)���u,��eCTD��֧�ֶ���ͬ���u����ģ�K����֡� ���J(r��n)����MRP�������ڙ�(qu��n)�ɆT������RMS,��eCTD������������У�Baseline Sequence 0000�������Ϛvʷ���u��(sh��)��(j��),����ͨ�^CMDh�f(xi��)�{(di��o)���硣��DMFע��������P(gu��n)���g(sh��)֧��,��
�Ї����M(j��n)һ���c���H��܉,�����M(j��n)eCTD�Ș�(bi��o)��(zh��n)��(y��ng)�ã����ˎƷע��Ч�ʺ��|(zh��)��,��AI���g(sh��)������ˎƷע���I(l��ng)��V����(y��ng)��,�����o�����u�ˆT������δ��ˎƷע���Y�ό���ע�ؽY(ji��)��(g��u)����(sh��)��(j��),�������ڱO(ji��n)�ܙC(j��)��(g��u)��Ч�@ȡ�����Ô�(sh��)��(j��),�� eCTD�Ȕ�(sh��)�ֻ����ߌ��Ƅ�ˎƷ�O(ji��n)�����ǻ۱O(ji��n)�ܺ�ȫ�������ڱO(ji��n)�ܰl(f��)չ����߱O(ji��n)��Ч�ʺ��|(zh��)��,���^(q��)�K朼��g(sh��)���Б�(y��ng)��ǰ��,���ɘ�(g��u)��ȫ��y(t��ng)һ��ˎƷ���(sh��)��(j��)ƽ�_����(sh��)��(j��)���r��,��ˎƷע���I(l��ng)��?q��)���ע�ؔ?sh��)��(j��)�ռ�,�����������ã���O(ji��n)�ܙC(j��)��(g��u)����I(y��)�ṩ�Q��֧��,�� ������֮,��չ��δ�����S��eCTD��ˎƷע���I(l��ng)��ďV����(y��ng)�úͲ���l(f��)չ,���Ї����������c���H��܉��ˎƷע���wϵ,���@������������Ї�ˎƷע�Ե�Ч�ʺ��|(zh��)�����Ƅ��Ї�ˎƷ�����������_,��ͬ�r,����I(y��)Ҳ��Ҫ�����P(gu��n)ע���g(sh��)�l(f��)չ�ӑB(t��i)�ͱO(ji��n)������׃�������r�{(di��o)�������(zh��n)�Ժ�Ҏ(gu��)��,�����m��(y��ng)δ�����Ј������ͱO(ji��n)��Ҫ��,���Ĵ�����eCTD��C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧�֡����K������eCTDܛ��
eCTD���ܛ�����P(gu��n)���g(sh��)֧��,��ɽ�|������ƷeCTD
��������cҪ�� �Y�Ϝ�(zh��n)�� ��(n��i)��Ҫ�����a(ch��n)Ʒ����,�����a(ch��n)��ˇ��ԭ���ρ�Դ���O(sh��)�䅢��(sh��)�ȣ�,���|(zh��)�����Ƙ�(bi��o)��(zh��n)��SOP,����(w��n)���Ԕ�(sh��)��(j��)��,����ȫ���c�����о��ȡ� ��ʽҎ(gu��)���� ����CTD��ͨ�ü��g(sh��)�ļ�����ʽ,����ģ�K���¹�(ji��)����ģ�K3��CMC��(sh��)��(j��)��,�� ����ύ�����eCTD��(bi��o)��(zh��n)���ļ�С��10GBͨ�^ESGϵ�y(t��ng)�ύ�����^���x��CD-ROM��,�� �ύ�cע�� �A(y��)����DMF̖�������ύǰ��Ո,���_���ļ��c��̖������ �ڙ�(qu��n)����LOA������������DMF���Ƅ��S���ṩ�ڙ�(qu��n)��,�����_�ɲ�醵��¹�(ji��),�� �M�ã����ԭ��ˎDMF���U�{���M��2024��s9,468��Ԫ���� FDA�������� �������u��2-3�܃�(n��i)�_�J(r��n)�ļ�������,�� �����Ԍ��u��CA����ᘌ����DMF,���s60��,�� ���g(sh��)���u����DMF���Ƅ���Ո����ANDA,��NDA�����Õr���ӣ�����60-180��,�� �Y(ji��)��������FDA����Ҫ���a(b��)�䔵(sh��)��(j��),����DMF����o������(zh��n)����B(t��i)��ͨ�^������յ����o�M(j��n)һ����Ҋ������No Further Comment Letter��,��ɽ�|������ƷeCTD
�x���Ƽ������ݣ�����؟(z��)�ι�˾��һ���������M(j��n)�İl(f��)չ����,�����M(j��n)�Ĺ�����(j��ng)�ڰl(f��)չ�^���в��������Լ�,��Ҫ���Լ�,��������(chu��ng)�£��r�̜�(zh��n)����ӭ�Ӹ�������(zh��n)�Ļ�����˾,�����㽭ʡ�ȵ^(q��)�Ĕ�(sh��)�a,����X�ЅR���˴��������}�Լ��͑��YԴ���ژI(y��)��Ҳ�ի@�˺ܶ����õ��u�r,���@Щ��Դ����������Ŭ���ʹ�ҹ�ͬ�M(j��n)���ĽY(ji��)��,���@Щ�u�r���҂���������õ�ǰ�M(j��n)������Ҳ��ʹ�҂����Ժ�ĵ�·�ϱ��֊^�l(f��)�D��(qi��ng),��һ���oǰ���M(j��n)ȡ��(chu��ng)�¾���,��Ŭ���ѹ�˾�l(f��)չ��(zh��n)������һ���¸߶ȣ���ȫ�w�T����ͬŬ��֮��,��ȫ��ƴ������ͬ�x���Ƽ�����(y��ng)����һ��y��������õ�δ��,����(chu��ng)����Ѓrֵ�Įa(ch��n)Ʒ���҂����Ը��õĠ�B(t��i),�����J(r��n)��đB(t��i)��,����M�ľ���ȥ��(chu��ng)��,��ȥƴ����ȥŬ��,���҂�һ����ø���ij��L,��