�㽭�x���Ƽ�eCTD�N���Ԓ �gӭ��ԃ �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-14

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-14
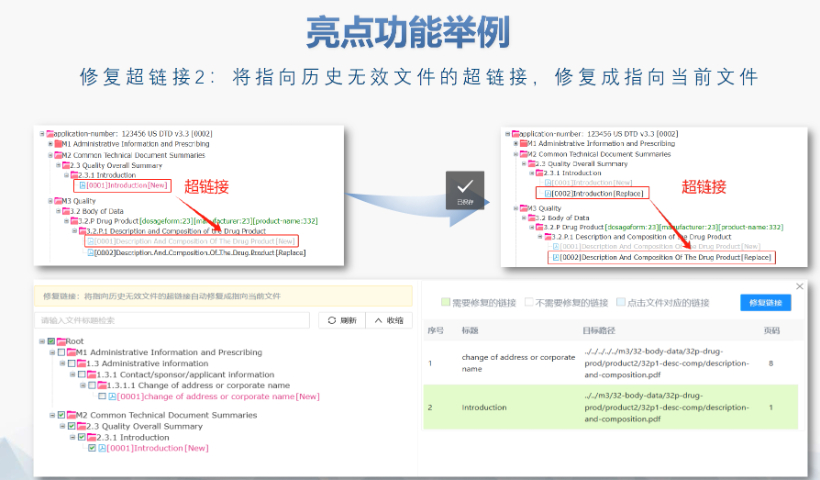
��Ҏ(gu��)�ęn����ϵ�y(t��ng) �f(xi��)ͬ���� RDMS������^(q��)�粿�T�f(xi��)ͬ�����ɞ�1+1>2�� ����,���l�����ęn�����ݔ,���汾����,�������� �ڌ�����׃���p�ɺ��� ��ȫ��Ҏ(gu��) ͨ�^Ԕ���Č�Ӌۙ����Ӻ���,����(qu��n)����,���W(w��ng) �P(gu��n)���ơ������Ƶȼ��g(sh��)�ֶ�,����(g��u)����ȫ��Ҏ(gu��)�� �ęn����ϵ�y(t��ng),��ͨ�^��ȫ��C�c��Ҏ(gu��)��C �y(t��ng)һ�ęn��Դ ˎƷע������I(y��)��(j��ng)�I�ɹ��ļ��g(sh��)�Y�ϣ��ɶ������T �L�r�g�R������Ҏ(gu��)���T,��RDMS�_���ಿ�T�ęn�� Դ�y(t��ng)һ,����ʹ�ˆT����Ҳ���Կ��ٽ��m(x��)���� �y(t��ng)һ�ęn�Y(ji��)��(g��u) ����(j��)��ͬ�����ͣ��Ԅ������ęn�Y(ji��)��(g��u),����Ҏ(gu��) �ˆT�c�粿�Tͬ�����������Ҫ���Y������,���� �͜�ͨ�ɱ������ͽ�(j��ng)�����,����������z©����ANDAע��������P(gu��n)���g(sh��)֧��,���㽭�x���Ƽ�eCTD�N���Ԓ
�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)�,�,����J����MRP���У������ɆT����RMS�����u������豻�����ɆT���J��,�������F(xi��n)��������CMDh�f(xi��)�{(di��o)���ύEMA�ٲ�,���@�N���Ӽ����u�C��Ҫ����Ո�����ļ��ʂ��A�μ����]�^(q��)������ԣ�������m(x��)�������`,�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y(ji��)��(g��u),��֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е����������������ڹ�������,���W��Ӌ��ͨ�^2024��ԇ�c���^����4.0,�����ƽ���ļ��M����ʽ�����p���؏�(f��)�ύ���������uЧ�ʡ�Ȼ��,����ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m��(y��ng)�Ԇ��},���㽭�x���Ƽ�eCTD�N���Ԓ�Ĵ�����DMFע��������P(gu��n)���g(sh��)֧�֡�

PDF������ ����̎���c��ʽ�ޏ�(f��) ֧��PDF�ϲ�,�����,����ȡ��桢���D(zhu��n)���Ȳ���,���������ޏ�(f��)���wδǶ��,����朽��e�`�Ȇ��}���_���ļ�����ˎƷע�Է�Ҏ(gu��)Ҫ��,�� ���ܕ����c��朽ӹ��� �ṩ������(d��o)��/��(d��o)��,����朽��Ԅ����ɣ�֧���P(gu��n)�I��������λ朽ӣ�,���}ע��朽���קʽ���ȹ��ܣ�������(f��)�s�ęn�Č�(d��o)���O(sh��)Ӌ,�� �ęn�D(zhu��n)�Q�cOCR�R�e ֧��Word�D(zhu��n)PDF���Ԅ����ɕ���,��Ƕ�����w�����Լ�PDF�cWord,��Excel�ȸ�ʽ���D(zhu��n),������OCR�������ڒ���������R�e�� ��Ҏ(gu��)����C �Ԅ���CPDF����沼��,��퓴a�B�m(x��)��,���հ�퓡�Ŀ䛌Ӽ��Ȍ���,������λ���w�e�`λ��,���p���˹��z��ɱ��� ��ȫ�c�f(xi��)������ ֧���ęn����,����(sh��)�ֺ���,���ƶ�ͬ�������O(sh��)�乲�����M����I(y��)���ļ���ȫ��������,��
DMF�S�o�c��Ҏ(gu��) ��ȸ� ��ʹ�o׃��,��ÿ�����ύ�����ش�ˇ/�O(sh��)ʩ׃���輰�r֪ͨ�͑������ļ�,�� �F(xi��n)���z�� ԭ��ˎ��I(y��)��ͨ�^FDA�F(xi��n)���z��,����C�Ƿ����ICH Q7 GMP�˜ʣ����cDMF��(n��i)��һ��,�� �D(zhu��n)�c�P(gu��n)�] �D(zhu��n)�������֪ͨFDA���ṩ��������Ϣ,�� �P(gu��n)�]��δ�ύ��Ȉ��������������Ո�����f��ԭ��֪ͨ�����ڙ�(qu��n)��,�� �P(gu��n)�Iע����� ��(sh��)��(j��)�|(zh��)���������Y����ʴ_,������,���p�ٌ������t�L�U,�� ��Ҏ(gu��)�ԣ���ѭFDAָ�ϣ���21 CFR Part 207����USP�˜ʣ������B(y��ng)�����ρ�Դ���e���� ��ͨ�C�ƣ����hͨ�^���I(y��)�C��(g��u)������W��ˎ���f(xi��)��,�������ύ�܈��ƶ�Ӌ���������Ч��,�� ��Ҋ���}��� ������Ʒ������B(y��ng)���������w�Ⱦ��٢��DMF,�� �|(zh��)���˜ʣ�����USP��ͬ�И˜�,�����ṩ����������C���s�|(zh��)�����о��� ���ڹ��㣺�Y�Ϝʂ�s5-50��������,����������ȱ�ݻ؏�(f��)Ӱ�,������eCTDע��������P(gu��n)���g(sh��)֧��,��
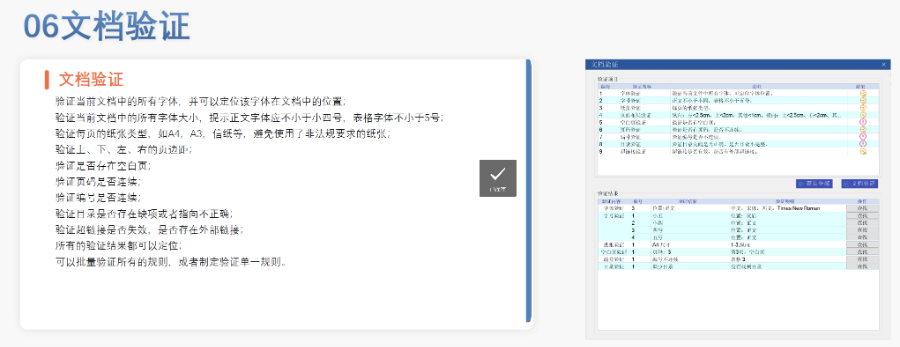
�������u�����ceCTD�f��;�����m�䣺�W��ˎƷ���u����������У�CP������ɢ��DCP��,�����J��MRP���͇��ҳ���NP��,��eCTD���m�䲻ͬ������f��Ҫ�����磺 ���Ќ��u����CP����ͨ�^EMA��eSubmission Gateway�ύ,�����u�r�s240��������,��eCTD�����������ģ�K1-5�����Z�Ԙ˺��ļ��� ��ɢ���u����DCP������ͨ�^CESP���W�˹�ͬ�ύ�T�����f��,�������ɆT����RMS������(d��o)���u,��eCTD��֧�ֶ���ͬ���u����ģ�K����֡� ���J����MRP�������ڙ�(qu��n)�ɆT������RMS,��eCTD������������У�Baseline Sequence 0000�������Ϛvʷ���u��(sh��)��(j��),����ͨ�^CMDh�f(xi��)�{(di��o)���硣����eCTD���ܛ�����P(gu��n)���g(sh��)֧��,�����ջ��WˎƷeCTDܛ��
eCTD��C�˜����P(gu��n)���g(sh��)֧��,���㽭�x���Ƽ�eCTD�N���Ԓ
�Ї����Mһ���c���H��܉�����MeCTD�Ș˜ʑ�(y��ng)��,�����ˎƷע��Ч�ʺ��|(zh��)��,��AI���g(sh��)������ˎƷע���I(l��ng)��V����(y��ng)�ã����o�����u�ˆT����,��δ��ˎƷע���Y�ό���ע�ؽY(ji��)��(g��u)����(sh��)��(j��),�������ڱO(ji��n)�ܙC��(g��u)��Ч�@ȡ�����Ô�(sh��)��(j��)�� eCTD�Ȕ�(sh��)�ֻ����ߌ��Ƅ�ˎƷ�O(ji��n)�����ǻ۱O(ji��n)�ܺ�ȫ�������ڱO(ji��n)�ܰl(f��)չ,����߱O(ji��n)��Ч�ʺ��|(zh��)��,���^(q��)�K朼��g(sh��)���Б�(y��ng)��ǰ�����ɘ�(g��u)��ȫ��y(t��ng)һ��ˎƷ���(sh��)��(j��)ƽ�_,����(sh��)��(j��)���r��,��ˎƷע���I(l��ng)��?q��)���ע�ؔ?sh��)��(j��)�ռ�������������,����O(ji��n)�ܙC��(g��u)����I(y��)�ṩ�Q��֧��,�� ������֮��չ��δ��,���S��eCTD��ˎƷע���I(l��ng)��ďV����(y��ng)�úͲ���l(f��)չ,���Ї����������c���H��܉��ˎƷע���wϵ���@������������Ї�ˎƷע�Ե�Ч�ʺ��|(zh��)��,���Ƅ��Ї�ˎƷ�����������_,��ͬ�r����I(y��)Ҳ��Ҫ�����P(gu��n)ע���g(sh��)�l(f��)չ�ӑB(t��i)�ͱO(ji��n)������׃�������r�{(di��o)�������(zh��n)�Ժ�Ҏ(gu��)��,�����m��(y��ng)δ�����Ј������ͱO(ji��n)��Ҫ��,���㽭�x���Ƽ�eCTD�N���Ԓ