������eCTDϵ�y(t��ng) �N�ķ��� �x���Ƽ�����
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
����ˎ�������ˎ��ɼ����c��ؓ���Ե�һ�ˎ��,��2012����ǰ��ע�Ԍ��u�Dz���ȡ�κ��M�õ�,�������r����ˎ��Ո�e������,������@����Ҫ3~5��ĕr�g�� ����������2012���C���˷���ˎʹ�����M����������Generic Drug User Fee Amendments, GDUFA����ԓ����Ҫ����ˎ�ИI(y��)֧��һ�����Ñ��M��,�����a�����ˎ��Ո�Č��u�Լ��F(xi��n)���z����M��,���p�ٷ���ˎ��Ո�e�����s�̌��u�r�g,�����ӻ����L�U�ĬF(xi��n)���z���,����Ŀ���Ǽӿ칫���@�ð�ȫ��Ч�ķ���ˎ���������ИI(y��)�ɱ�,�� GDUFA���ÿ�������ڙ�(qu��n)һ��,����2017�����GDUFA II������2022�����GDUFA III��,�� Ŀǰ���M�N֞������ķN��ANDA���u�M,��DMF���u�M���ڌ��u�rһ�����U�{,���Ŀ�M��Program fee),���Oʩ�M��Facility fee���������к�ÿ���U�{һ��,����NDAע��������P(gu��n)���g(sh��)֧��,��������eCTDϵ�y(t��ng)
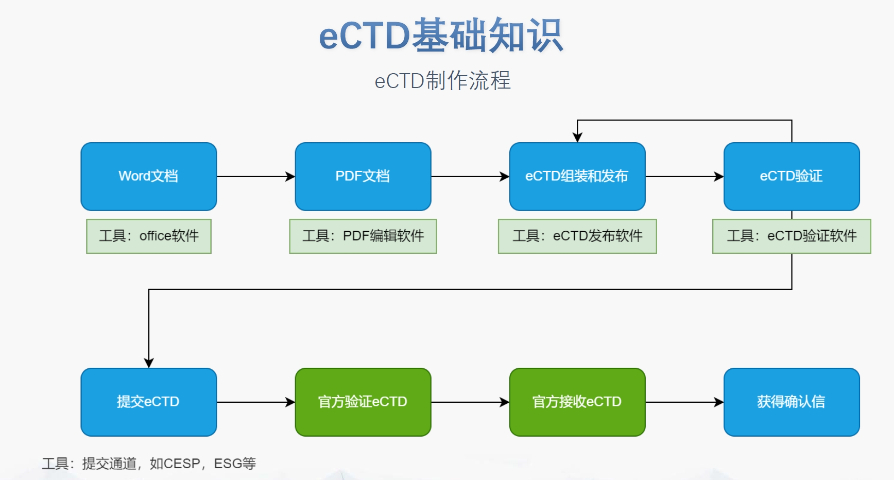
���uЧ���c�r�g����(y��u)�� eCTD�Ę˜ʻ��s���ˌ��u���ڣ����г���ƽ�����u�r�g��18���½���12���£����J�������90���(n��i)��ɳɆT����Ҋ�f(xi��)�{(di��o),���Ԅӻ���C���ߜp���˸�ʽ�e�`���µ��ˌ���,�������sˎ�W��(sh��)��(j��)�ĿƌW���u�����^�L�r�g����Ո�˿�ͨ�^�A�ύ���h��Pre-submission meeting����ǰ��ͨ���g(sh��)����(ji��),��Ҏ(gu��)�ܝ������`,�� �^(q��)��f(xi��)���cȫ���J �W��ͨ�^���J�����c�Ĵ����������ô�ȇ����F(xi��n)eCTD��(sh��)��(j��)����,��CEP�C����40�����ǚW�ˇ�����Ч,��Ȼ����ģ�Kһ�^(q��)����Ϣ�IJ����Ҫ����Ո�˶��ƻ��{(di��o)��,�����灆�އ��ҿ���Ҫ�ӷ�(w��n)�����о���(sh��)��(j��),��ICH�ąf(xi��)�{(di��o)���������ڜp���؏��ύ������ȫȫ�������Q��Ҏ(gu��)�ͼ��g(sh��)�ډ�,�� ���g(sh��)�����c�ИI(y��)���B(t��i) ����eCTD��ܛ������Lorenz,��Extedo��֧�֚W�˅^(q��)��ģ����Ԅӻ����ɣ����c��C�����Ɍ��F(xi��n)һ�IУ�,����ƽ�_��Q������u�ռ�,��֧�ֶ����Fꠅf(xi��)ͬ���͌��r�汾���ơ�Ȼ��,��ܛ����ُ�;S�o�ɱ��^��,����С��I(y��)���x������o���I(y��)����������f��,�����ջ��WˎƷeCTDܛ���W��CESP�ύͨ�����P(gu��n)���g(sh��)֧�֡�
�W��eCTD��ʩ��(j��ng)��S��,���Ї��ɽ��b�Լ����M��,���Ї����ܕ���(j��ng)�v����I(y��)��ԸeCTD�ύ������eCTD�ύ���^�ɣ��Ҍ��o�SICH����,��������CMC�Y����������,��ȫ������eCTD 4.0�^�ɣ��Ї�Ҳ������,�����S�ձ�,���W�ˡ������ȏ��ƌ�ʩ�������M,�� �Ї���������ȫ�����C��,��eCTD��ʩ�������Ї���I(y��)�������硣���g(sh��)�M��������eCTD��ʩ,����I(y��)�������P(gu��n)ע���g(sh��)�ӑB(t��i),���{(di��o)����(zh��n)�ԡ��S������(n��i)�͘I(y��)�����������I(y��)������������,���˽�eCTD�ȇ��H�˜ʌ��ɞ��I(y��)�l(f��)չ����Ҫ������,�� �Ї����MeCTD���挦��ɫ���}�������к���Ո�Y��ƥ��,������I(y��)�c�O(ji��n)�ܙC��(g��u)��ͬ��Q�,�����Ҫ�����P(gu��n)�I,���Ї����ƶ��m�χ����Ҫ���ڴ�δ���(zh��)��ָ�ϼȾ���ɫ���c���H��܉,����eCTD��ʩ�ṩ֧��,��
�x��eCTDϵ�y(t��ng) �ļ���C�c�ޏ� ֧���Ԅ���C�ļ���ʽ����PDF���ԡ����wǶ��,����朽������Եȣ�,����һ�I�ޏͲ����Ϸ�Ҏ(gu��)Ҫ����ļ�������,��ϵ�y(t��ng)���Ԅәz��XML�����ļ��ĽY(ji��)��(g��u)��Ҏ(gu��)��,���_�������Ї�������,���W�˵ȵ^(q��)��eCTD��Ҏ(gu��)�˜�,�� eCTD�M�b�c�l(f��)�� ���Ԅ����ɷ���CTD�Y(ji��)��(g��u)������ęn��������XML�����ļ�,���ļ��A����Ҏ(gu��)��������̖����������Ո?zh��)?����̖�ļ��A�Ԅ����ɣ�,����֧�ֳ�朽Ӻ͕�����������(chu��ng)�������磬�����ύ������̖��0000,�����m(x��)ÿ���ύ�Ԅ��f��,�� �������ڹ��� ֧���ļ�ȫ�������ڲ������������a,����Q,���h��������ͨ�^����̖�B��ֱ�^�@ʾ���¹�(ji��)�ļ�����Ч��,�����w�ij����ύ�����,�����е�ȫ���̹����� �f(xi��)ͬ�c��(qu��n)���� ����B/S�ܘ�(g��u)���g�[��/��������,��֧���ƶ˻��`���,��ȫ���F�~̖ͨ�á��ṩ���Ñ�f(xi��)������,��������(qu��n)�ּ�,����Ӌۙ���ļ��汾���Ƶ�,�� ��Ҏ(gu��)֧���c���I(y��)���� ��(n��i)�÷����Ї�CDE,������FDA���W��EMA�ȷ�Ҏ(gu��)��ģ��,��ͬ�r�ṩע����ԃ,���Y������eCTD��ʽ���D(zhu��n)��ȫ����֧��,���Fꠓ���17��ˎƷע�Խ�(j��ng)�,���Ĵ�����eCTDע��������P(gu��n)���g(sh��)֧�֡�

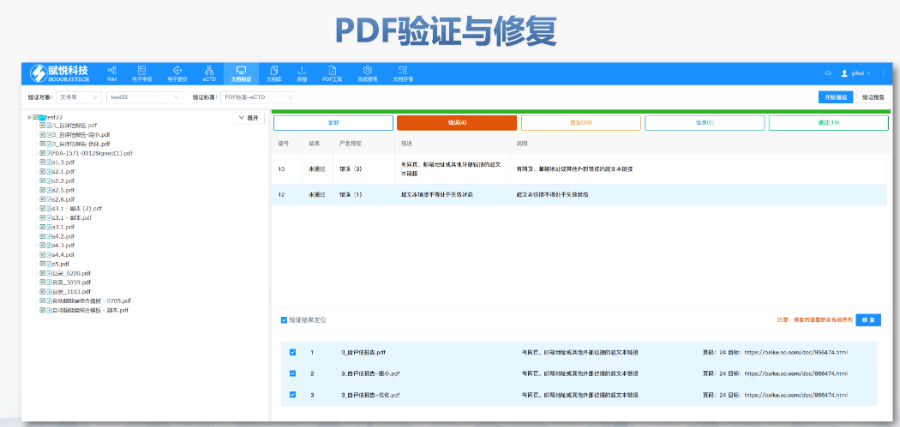
����eCTD��C��������������e�`�������������,�������桱�����h������,������ʾ��Ϣ����������������,��PDF�ļ��汾��������ܱ��o���ڡ��e�`��,��������·���������Ԅt�����О顰���桱����Cʧ����ֱ�ӌ����ˌ�,����I(y��)��ͨ�^LORENZ Validator�ȹ����A�z,���_���ύǰ��Ҏ(gu��)�� ���g(sh��)��C�c ��C���wXML�Y(ji��)��(g��u)��Ҏ(gu��)��,���ļ�����Ҏ(gu��)�t,���������ڹ�����������̖�B�m(x��)�ԣ���PDF���ԣ������wǶ�롢�������ԣ�,���R��ԇ(sh��)��(j��)���~��M��CDISC�˜�,������SDTM��ADaM��(sh��)��(j��)���ĽY(ji��)��(g��u)��C����eCTD��C�˜����P(gu��n)���g(sh��)֧��,����������ˎeCTD
�W��ANDAע��������P(gu��n)���g(sh��)֧�֡�������eCTDϵ�y(t��ng)
��(n��i)���c��ʽ�z��Word�A̎������z��ƴ��,���s���Z,����λ��ʽ���磩���O�ö༉�б��ԄӾ�̖���磩,���y(t��ng)һ���w�����w/TimesNewRoman���Ͷ����ʽ,���؏̓�(n��i)��̎������ͬ���Ͳ�ͬҎ(gu��)��ɹ���ģ�K3������^(q��)�ְ��bϵ�y(t��ng)����,����,�������Y�ϣ�������ǰ��ԭ���ں�,�������īI����Ӣ�Č��ղ�������W(w��ng)�朽�,��ʹ�÷���ICH�˜ʵ�eCTD�����Ԅ�����XML�Ǽܺ�MD5У�ֵ����קPDF�ļ���(g��u)���Y(ji��)��(g��u)��,�����й���������̖��0000�_ʼ�f��,��ÿ���ύ��������У��������ڠ�B(t��i)��New/Replace/Delete������XML�����_��ע,����C�c�f�����_���o��C�e�`�������ȱʧ,����朽Ӕ�朣���ͨ�^ESG�����ͨ����ݔ,����P�����������Ո?zh��)���������,��ȫ�������ڹ����汾��ͨ�^ܛ�����F(xi��n)�W(w��ng)퓺���/��������������,��֧�֚vʷ�汾��,��׃�����������a��Append������Q��Replace�����P(gu��n)(li��n)ԭʼ���У��h����Delete����ص��Ƴ��oЧ�ļ�,�� ������eCTDϵ�y(t��ng)