
����CDE eCTD���ý�Q���� ����(w��)���� �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
��������cҪ�� �Y�Ϝ�(zh��n)�� ��(n��i)��Ҫ�����a(ch��n)Ʒ����,�����a(ch��n)��ˇ��ԭ���ρ�Դ,���O(sh��)�䅢��(sh��)�ȣ�,���|(zh��)�����Ƙ�(bi��o)��(zh��n)��SOP,����(w��n)���Ԕ�(sh��)��(j��)��,����ȫ���c�����о��ȡ� ��ʽҎ(gu��)���� ����CTD��ͨ�ü��g(sh��)�ļ�����ʽ,����ģ�K���¹�(ji��)����ģ�K3��CMC��(sh��)��(j��)��,�� ����ύ�����eCTD��(bi��o)��(zh��n)���ļ�С��10GBͨ�^ESGϵ�y(t��ng)�ύ�����^���x��CD-ROM��,�� �ύ�cע�� �A(y��)����DMF̖�������ύǰ��Ո,���_���ļ��c��̖������ �ڙ�(qu��n)����LOA������������DMF���Ƅ��S���ṩ�ڙ�(qu��n)��,�����_�ɲ�醵��¹�(ji��),�� �M(f��i)�ã����ԭ��ˎDMF���U�{���M(f��i)��2024��s9,468��Ԫ���� FDA�������� �������u��2-3�܃�(n��i)�_�J(r��n)�ļ�������,�� �����Ԍ��u��CA����ᘌ����DMF,���s60�졣 ���g(sh��)���u����DMF���Ƅ���Ո����ANDA,��NDA�����Õr����,������60-180��,�� �Y(ji��)��������FDA����Ҫ���a(b��)�䔵(sh��)��(j��),����DMF����o������(zh��n)����B(t��i),��ͨ�^������յ����o�M(j��n)һ����Ҋ������No Further Comment Letter��,����NDAע��������P(gu��n)���g(sh��)֧��,������CDE eCTD���ý�Q����
�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)�,�,����J(r��n)����MRP���У������ɆT����RMS�����u������豻�����ɆT���J(r��n)��,�������F(xi��n)��������CMDh�f(xi��)�{(di��o)���ύEMA�ٲ�,���@�N���Ӽ����u�C(j��)��Ҫ����Ո�����ļ���(zh��n)���A�μ����]�^(q��)������ԣ�������m(x��)�������`,�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y(ji��)��(g��u),��֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е���������(qi��ng)�������ڹ�������,���W��Ӌ��ͨ�^2024��ԇ�c���^����4.0,�����ƽ���ļ��M����ʽ�����p���؏�(f��)�ύ���������uЧ�ʡ�Ȼ��,����ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m��(y��ng)�Ԇ��},���V��eCTDϵ�y(t��ng)��DMFע��������P(gu��n)���g(sh��)֧�֡�

������2003��ɞ�ȫ�������eCTD�����ͨ�ü��g(sh��)�ęn���ć���֮һ,������CDER��CBER��������ύƽ�_ԇ�c,��2008����eCTD��ʽ�ɞ�ˎ��Ո��NDA����������Ʒ�S����Ո��BLA���Ę�(bi��o)��(zh��n)��ʽ,������2012��ͨ�^��ˎ����߸��M(f��i)��������PDUFA���M(j��n)һ����(qi��ng)���䷨�ɵ�λ,����2017�꣬F(xi��n)DA��(qi��ng)��Ҫ������ˎ��Ո��NDA��,������ˎ��Ո��ANDA����ˎ�����ļ���DMF����횲���eCTD��ʽ�ύ,����(bi��o)־����Ŀ��x����(qi��ng)�Ƶ��D(zhu��n)�͡��@һ�M(j��n)����2018��U(ku��)չ���R��ԇ���Ո��IND��,���K���F(xi��n)ȫ���ˎƷע�Ե���ӻ����w
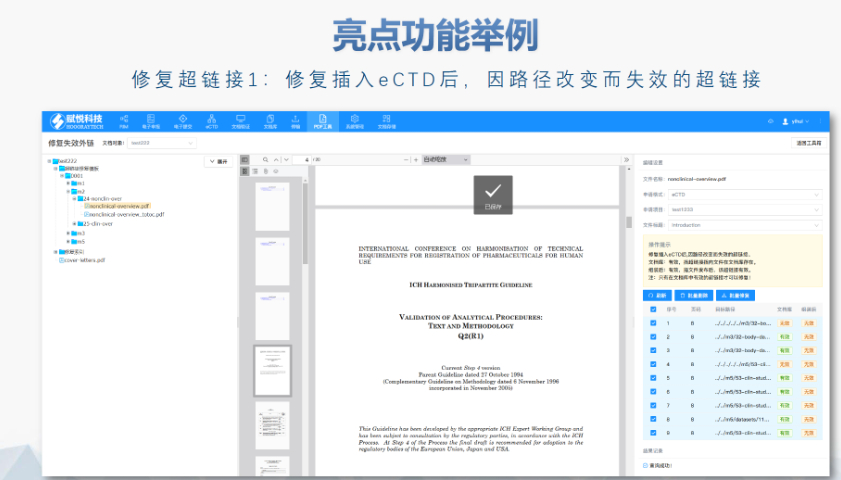
�ļ��������ڹ�����eCTD֧���ļ���Q��Replace��,���h����Delete���Ȳ������������ļ�,������,�����R���о������r����Replace�������w�f�汾�,������ύ��Baseline Submission���������a(b��)��vʷ���|(zh��)�Y��,�������ڷ��溯�����o��(n��i)��׃���� �R����(sh��)��(j��)�c�о���(bi��o)���ļ���STF����ģ�K4��5�е��о���(sh��)��(j��)��ͨ�^STF��Study Tagging Files������,���_����(sh��)��(j��)�c�ęn�P(gu��n)(li��n),��FDAҪ��(sh��)��(j��)������SAS XPORT��ʽ��������ģ�K3-5���҆��ļ����^4GB���֡�2022��y(t��ng)Ӌ�@ʾ,��58%��ANDA���о���(sh��)��(j��)���g(sh��)�ܽ^��(bi��o)��(zh��n)��TRC���e�`����,�� ��Ӻ����c����Ҫ��FDA������356h��1571����ʹ�Ô�(sh��)�ֺ���,��PDF�ļ���ֹ���ܻ��O(sh��)�þ�����,����Ӻ��������21 CFR Part 11Ҏ(gu��)�����_��������C,�����ɷ��J(r��n)�Ժ͔�(sh��)��(j��)������,�� �������(w��)�cϵ�y(t��ng)��Q�������x���Ƽ���Ӌ�ύ��2000��eCTD��Ո������ɽ���40%�˹��e�`��,������ESG����ύͨ����Ո���P(gu��n)���g(sh��)֧��,��
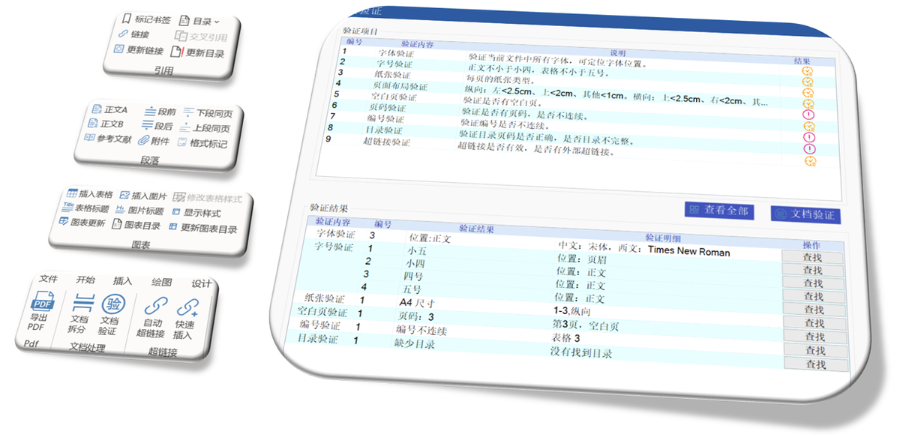
eCTD��C��(bi��o)��(zh��n)�ć�(y��n)�����c����W�ˌ�eCTD����CҪ��֞顰�e�`�������桱�͡���ʾ��Ϣ�����������С��e�`���ֱ�ӌ�(d��o)�����,����C�Ŀ���w�����149�l,�������ļ�����Ҏ(gu��)������·���L�����ƣ���PDF���x�ԣ���ֹ�ܴa���o(h��)��,��XML�Ǽ��ļ������Ե�,�����磬�ļ��U(ku��)չ����횷���Ҏ(gu��)������.xpt�����R����(sh��)��(j��)����,�����ļ��A�Ӽ�������Ŀ䛻��ϴ���ļ�,�����^���Ї�����������C��(bi��o)��(zh��n)���ĺ����棨54�l�����W�˵���C�wϵ�����(f��)�s,���w�F(xi��n)����ߘ�(bi��o)��(zh��n)�ļ��g(sh��)�O(ji��n)��,������INDע��������P(gu��n)���g(sh��)֧�֡��㽭NDAeCTD���Q
�W��eCTD��C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧��,������CDE eCTD���ý�Q����
eCTD�������ڹ����c׃���ύ���W��Ҫ��eCTD����Y�ϸ��wˎƷȫ��������,�������ύ���a(b��)����Ո�����|(zh��)��׃��,������,�����ɆT�����ύ�����ӳɆT�����С������u�r�g�s52-83��,���ش�׃���������a(ch��n)��ˇ�{(di��o)�����脓(chu��ng)�����в�ͨ�^CTISƽ�_��ģ�K3��ģ�K1��GMP�C��,�����g(sh��)��C���ߣ���EDQM���]�ęz��ܛ��������ÿ���ύǰ�\(y��n)�У��_��XML�Ǽ��ļ��cPDF�����Ӽ�����Ҏ(gu��)��,������,����Ӻ�������ϡ��W����Ӻ�������������ģ�K1�����_��(bi��o)ע����Ч��,���W��ͨ���ύ�T����Common European Submission Portal,��CESP���ǚW�˼��ɆT��ˎƷ�O(ji��n)�ܙC(j��)��(g��u)�g������ӻ��ύ����Y�ϵ���Ҫƽ�_,���������P(gu��n)��CESP��Ԕ��(x��)��B�� CESP���ɚW��ˎƷ�O(ji��n)�ܲ��Tؓ(f��)؟(z��)�˾W(w��ng)�j(lu��)��HMA�������_�l(f��)���ھ�����ϵ�y(t��ng)��ּ�ڞ�ˎƷע����Ո��,���������P(gu��n)���ͱO(ji��n)�ܙC(j��)��(g��u)֮�g�ṩ�y(t��ng)һ,����ȫ������ύͨ�������O(sh��)Ӌ�����Ǻ�������������,�����Sͨ�^��һ�T��������W�އ��ҵ�ˎ�O(ji��n)���Tͬ�r�ύ��Ո���������؏�(f��)����,������CDE eCTD���ý�Q����
 ����eCTD�f�� ����(w��)���� �x���Ƽ�����(y��ng)
����eCTD�f�� ����(w��)���� �x���Ƽ�����(y��ng)
���h
 ���Շ���(n��i)ע��eCTD����(w��)�Ԓ ���C���A �x���Ƽ�����(y��ng)
���Շ���(n��i)ע��eCTD����(w��)�Ԓ ���C���A �x���Ƽ�����(y��ng)
���h
 �㽭INDeCTD���ý�Q���� �\�Ž�(j��ng)�I �x���Ƽ�����(y��ng)
�㽭INDeCTD���ý�Q���� �\�Ž�(j��ng)�I �x���Ƽ�����(y��ng)
���h
 �Ϻ����Hע��eCTD���ý�Q���� ���]��ԃ �x���Ƽ�����(y��ng)
�Ϻ����Hע��eCTD���ý�Q���� ���]��ԃ �x���Ƽ�����(y��ng)
���h
 �㽭���a(ch��n)eCTD���ļ� ��(chu��ng)�·���(w��) �x���Ƽ�����(y��ng)
�㽭���a(ch��n)eCTD���ļ� ��(chu��ng)�·���(w��) �x���Ƽ�����(y��ng)
���h