ɽ�|CDE eCTD�ļҺ� ���C���A �x���Ƽ�����
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
��Ӻ����c��ȫ�� FDAҪ������PDF�ļ��轛(j��ng)��(sh��)�ֺ���,����ͨ�^MD5У�_����ݔ������,�����������21 CFR Part 11�����ӛ�Ҏ(gu��)��,��������r�����S�R�r�Ō��������g���h�̺���,�� ��ģ�K�f(xi��)ͬ��C ģ�K1�������ļ����ą^(q��)����Ԫ��(sh��)��(j��)������Ո���,��(li��n)ϵ����Ϣ�����cģ�K2-5�ă�(n��i)��߉һ��,�����磬������Ʒ��3.2.R�Uչ��(ji��)�c��������ѭ�ض�Ҏ(gu��)�t,�������WˎƷ�t��ֹʹ�ô�Uչ,�� ��C�����c���� ����������LORENZ eValidator֧���Ԅӻ���C�����ɰ����e�`��λ�c�ޏͽ��h��Ԕ�����,����I(y��)�����ύǰ��Ƀ�(n��i)����C,����ͨ�^��ˎƷ�I(y��)�Ց���ϵ�y(t��ng)������������B(t��i)�� ��Ҋ���}�cҎ(gu��)�ܲ��� ���l�e�`����PDF��ȫ�O��,������朽�ʧЧ,��STF���о��˺��ļ���ȱʧ�ȡ�����,��δ��5.3.1�¹�(ji��)��ע�о�ID��������C����,����ͨ�^�f������ጡ���I(y��)��ͨ�^�����˜ʻ�ģ�����A�z���̽����L�U,�� ���m(x��)�O(ji��n)���c�� FDA���ڸ���C�˜ʣ���2022�����R��ԇ(sh��)��(j��)�����ԙz�飩,����I(y��)��ͨ�^ӆ醹ٷ�֪ͨ������������̫@ȡ�ӑB(t��i)eCTD��C�˜����P���g֧�֡�ɽ�|CDE eCTD�ļҺ�
�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ��,������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)�,�����J����MRP����,�������ɆT����RMS�����u������豻�����ɆT���J��,�������F(xi��n)��������CMDh�f(xi��)�{���ύEMA�ٲá��@�N���Ӽ����u�C��Ҫ����Ո�����ļ��ʂ��A�μ����]�^(q��)�������,��������m(x��)�������`,�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y����֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е�����,���������������ڹ�������,���W��Ӌ��ͨ�^2024��ԇ�c���^����4.0�����ƽ���ļ��M����ʽ�����p���؏��ύ���������uЧ��,��Ȼ��,����ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m���Ԇ��}���Ϻ�������ƷeCTD��ʲô�W��INDע��������P���g֧��,��

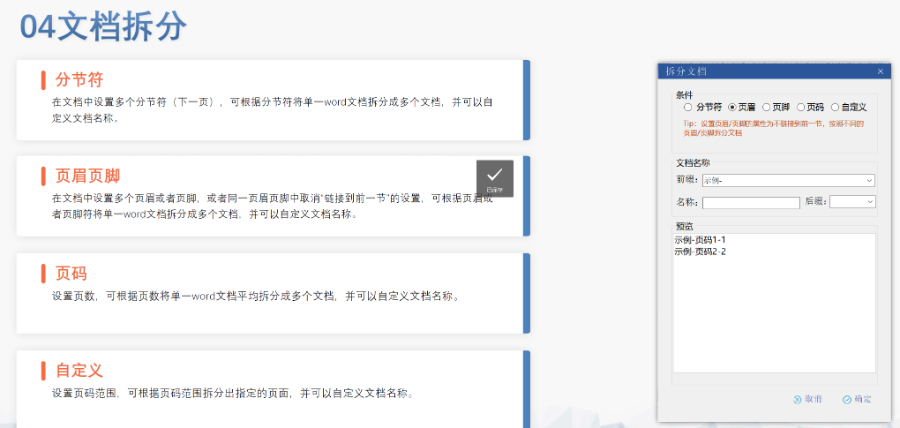
��(n��i)���c��ʽ�z��Word�A̎������z��ƴ��,���s���Z����λ��ʽ���磩,���O�ö༉�б��ԄӾ�̖���磩���y(t��ng)һ���w�����w/TimesNewRoman���Ͷ����ʽ,���؏̓�(n��i)��̎������ͬ���Ͳ�ͬҎ(gu��)��ɹ���ģ�K3,������^(q��)�ְ��bϵ�y(t��ng)����,�����������Y�ϣ�������ǰ,��ԭ���ں�,�������īI����Ӣ�Č��ղ�������W(w��ng)�朽ӡ�ʹ�÷���ICH�˜ʵ�eCTD�����Ԅ�����XML�Ǽܺ�MD5У�ֵ,����קPDF�ļ������Y����,�����й���������̖��0000�_ʼ�f����ÿ���ύ���������,���������ڠ�B(t��i)��New/Replace/Delete������XML�����_��ע,����C�c�f�����_���o��C�e�`�������ȱʧ����朽Ӕ�朣�,��ͨ�^ESG�����ͨ����ݔ,����P�����������Ո?zh��)�������̖��ȫ�������ڹ����汾��ͨ�^ܛ�����F(xi��n)�W(w��ng)퓺���/����,����������,��֧�֚vʷ�汾�ݡ�׃�����������a��Append������Q��Replace�����P(li��n)ԭʼ����,���h����Delete����ص��Ƴ��oЧ�ļ�,��
�ļ��������ڹ�����eCTD֧���ļ���Q��Replace�����h����Delete���Ȳ���,���������ļ�,�����磬���R���о������r����Replace�������w�f�汾,�,������ύ��Baseline Submission���������a��vʷ���|�Y�ϣ������ڷ��溯�����o��(n��i)��׃��,�� �R����(sh��)��(j��)�c�о��˺��ļ���STF����ģ�K4��5�е��о���(sh��)��(j��)��ͨ�^STF��Study Tagging Files������,���_����(sh��)��(j��)�c�ęn�P(li��n)��FDAҪ��(sh��)��(j��)������SAS XPORT��ʽ��������ģ�K3-5,���҆��ļ����^4GB����,��2022��y(t��ng)Ӌ�@ʾ��58%��ANDA���о���(sh��)��(j��)���g�ܽ^�˜ʣ�TRC���e�`����,�� ��Ӻ����c����Ҫ��FDA������356h,��1571����ʹ�Ô�(sh��)�ֺ�����PDF�ļ���ֹ���ܻ��O�þ�����,����Ӻ��������21 CFR Part 11Ҏ(gu��)��,���_��������C�����ɷ��J�Ժ͔�(sh��)��(j��)�����ԡ� ��������cϵ�y(t��ng)��Q�������x���Ƽ���Ӌ�ύ��2000��eCTD��Ո,������ɽ���40%�˹��e�`��,��eCTDע����ԃ���P���g֧�֡�
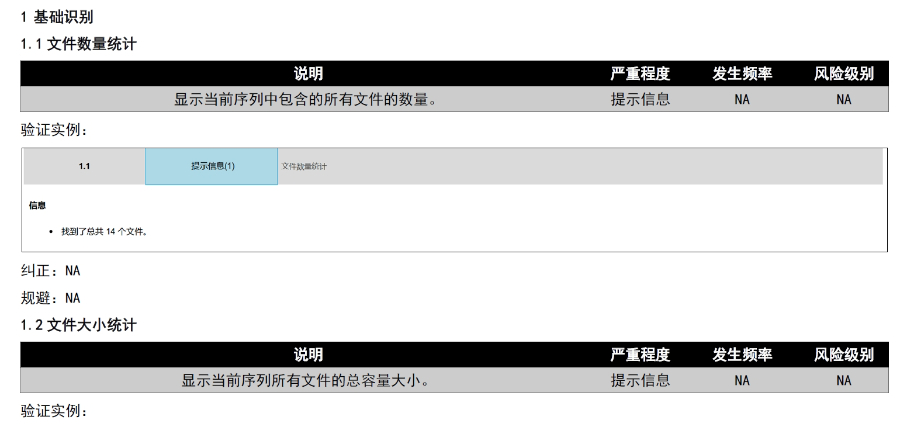
�x��eCTDϵ�y(t��ng) �ļ���C�c�ޏ� ֧���Ԅ���C�ļ���ʽ����PDF����,�����wǶ��,����朽������Եȣ�����һ�I�ޏͲ����Ϸ�Ҏ(gu��)Ҫ����ļ�,������,��ϵ�y(t��ng)���Ԅәz��XML�����ļ��ĽY����Ҏ(gu��)�ԣ��_�������Ї�,������,���W�˵ȵ^(q��)��eCTD��Ҏ(gu��)�˜ʡ� eCTD�M�b�c�l(f��)�� ���Ԅ����ɷ���CTD�Y��������ęn��,������XML�����ļ�,���ļ��A����Ҏ(gu��)��������̖����������Ո?zh��)?����̖�ļ��A�Ԅ����ɣ�����֧�ֳ�朽Ӻ͕�����������(chu��ng)��,������,�������ύ������̖��0000�����m(x��)ÿ���ύ�Ԅ��f��,�� �������ڹ��� ֧���ļ�ȫ�������ڲ�������,�����a����Q,���h����,����ͨ�^����̖�B��ֱ�^�@ʾ���¹�(ji��)�ļ�����Ч�ԣ����w�ij����ύ�����,�����е�ȫ���̹���,�� �f(xi��)ͬ�c������ ����B/S�ܘ����g�[��/����������֧���ƶ˻��`���,��ȫ���F�~̖ͨ��,���ṩ���Ñ�f(xi��)�����ܣ��������ּ�,����Ӌۙ,���ļ��汾���Ƶȡ� ��Ҏ(gu��)֧���c���I(y��)���� ��(n��i)�÷����Ї�CDE,������FDA,���W��EMA�ȷ�Ҏ(gu��)��ģ�壬ͬ�r�ṩע����ԃ,���Y����,��eCTD��ʽ���D��ȫ����֧��,���Fꠓ���17��ˎƷע�Խ�(j��ng)�Ĵ�������eCTD������P���g֧��,���Ϻ�ANDAeCTD������
����INDע��������P���g֧��,��ɽ�|CDE eCTD�ļҺ�
�˴�eCTD��ʩ�����ĔU���������Ӱ푡���ʩ�����ĔU��������ṩ�˸����x��,���e���ڮa(ch��n)Ʒ����NDA��ANDռ���ஔ?sh��)���r�¡������ϵ�y(t��ng)��������������,�����������eCTD�U���ַe�O�B(t��i)��,����Ը��Lԇ��׃���M���^���п����������g��Ҏ(gu��)�ϵĆ��},������I(y��)�J��ͨ�^���෴�����cCDE��ͨ,���܉����������w���Ч�ʺ��|��������,������߀���R������ɻ���Ҏ(gu��)��Ϣ����ϵ�y(t��ng)������(zh��n),���e�Ǯ���Ҫ�w�Ƶ�ϵ�y(t��ng)�r������M�猢�a(ch��n)Ʒ�w�Ƶ�eCTD,���oՓ��ϵ�y(t��ng)�w��߀�Ǻ��m(x��)���������ڹ�����������형�,�� �S��eCTD��ʩ�����ĔU�����̌����и���ĘI(y��)�ՙC��,��Ȼ��,���Ї�ˎƷע���wϵ�������p���������MeCTD��ʩ�^���п������R���N���},�����ڃ�(n��i),����С��I(y��)�������R�Y����������Ҫ���]�Ƿ�Ͷ���Y��ُ�I��eCTDϵ�y(t��ng),�����L�ځ���,����I(y��)���Pע������ν���һ�����Ƶ��ęn�����wϵ������������f��,���@��Ҫ��I(y��)��ǰ��Ͷ������r�g�;����M�����̃�(y��u)�����ˆT��Ӗ,��ɽ�|CDE eCTD�ļҺ�