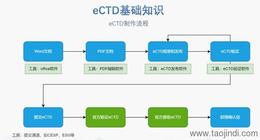
�㽭����ˎeCTD����(w��)�Ԓ ��(l��i)���ԃ �x���Ƽ�����(y��ng)
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-30

�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-30
�W��eCTD��(sh��)ʩ��(j��ng)�(y��n)�S�����Ї�(gu��)�ɽ��b�Լ����M(j��n)��,���Ї�(gu��)���ܕ�(hu��)��(j��ng)�v����I(y��)��ԸeCTD�ύ����(qi��ng)��eCTD�ύ���^(gu��)��,���Ҍ��o�SICH������������CMC�Y����������,��ȫ������eCTD 4.0�^(gu��)��,���Ї�(gu��)Ҳ�����⣬���S�ձ�,���W��,������(gu��)�ȏ�(qi��ng)�ƌ�(sh��)ʩ�������M(j��n)�� �Ї�(gu��)��������(l��i)ȫ��(j��ng)��(zh��ng)�C(j��)��(hu��),��eCTD��(sh��)ʩ�������Ї�(gu��)��I(y��)��������,�����g(sh��)�M(j��n)��������eCTD��(sh��)ʩ����I(y��)�������P(gu��n)ע���g(sh��)��(d��ng)�B(t��i),���{(di��o)����(zh��n)��,���S����(gu��)��(n��i)�͘I(y��)�����������I(y��)�����������ӣ��˽�eCTD�ȇ�(gu��)�H��(bi��o)��(zh��n)���ɞ��I(y��)�l(f��)չ����Ҫ��(j��ng)��(zh��ng)��,�� �Ї�(gu��)���M(j��n)eCTD���挦(du��)��ɫ��(w��n)�},�������к���Ո(q��ng)�Y��ƥ�䣬����I(y��)�c�O(ji��n)�ܙC(j��)��(g��u)��ͬ��Q,�,�����Ҫ�����P(gu��n)�I���Ї�(gu��)���ƶ��m�χ�(gu��)���Ҫ��,���ڴ�δ��(l��i)��(zh��)��ָ�ϼȾ���ɫ���c��(gu��)�H��܉,����eCTD��(sh��)ʩ�ṩ֧�֡��W��NDAע��(c��)���(b��o)���P(gu��n)���g(sh��)֧��,���㽭����ˎeCTD����(w��)�Ԓ
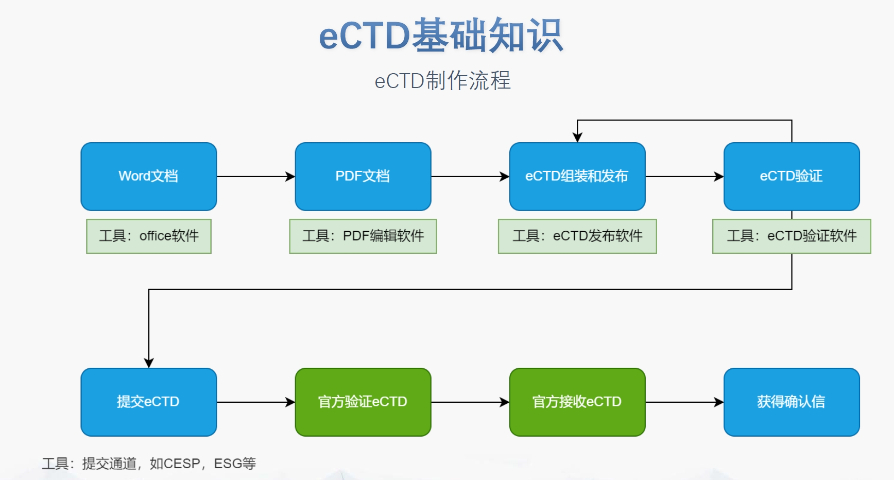
��(j��ng)��(j��)Ӱ��c�ɱ�Ч�� �M�ܳ���Ͷ���^�ߣ�ƽ��ÿ��I(y��)��50�f(w��n)�WԪ��,����eCTD�ɜp��30%�Č��u(p��ng)���t�ɱ����L(zh��ng)��Ч��,������ˎ��I(y��)ͨ�^(gu��)eCTD��(f��)��ԭ�Д�(sh��)��(j��),����(ji��)ʡ80%�����(b��o)��(zh��n)��r(sh��)�g,���W���A(y��)��ܿ�2�|�WԪ�Y����С��I(y��)��ɔ�(sh��)�ֻ��D(zhu��n)�͡� ���팏���c��(sh��)��(j��)�[˽ eCTD�еĻ��ߔ�(sh��)��(j��)��������̎��,�����ϡ�ͨ�Ô�(sh��)��(j��)���o(h��)�l������GDPR��Ҫ��,���R��ԇ�(y��n)?z��i)��K��ģ�K5�����ύ�踽������ί�T��(hu��)����(zh��n)�ļ����҅^(q��)��汾���w�F(xi��n)����(gu��)���팏��,��AI�o�������������ڱ��o(h��)�[˽��ͬ�r(sh��)������(sh��)��(j��)̎��Ч��,�� ���g(sh��)�ں��c���I(l��ng)��(y��ng)�� eCTD��ʽ�U(ku��)չ���t(y��)����е�ͱ���Ʒ�I(l��ng)�W��ԇ�c(di��n)eCTD-MDR�(xi��ng)Ŀ����ISO��(bi��o)��(zh��n),�,�����a(ch��n)Ʒ��eCTD�踽�����ﰲȫ��(sh��)��(j��)��(k��)�����c�W�˻����(k��)��(sh��)�r(sh��)ͬ��,��δ��(l��i),��eCTD���c��ӽ����n����EHR��ϵ�y(t��ng)��(du��)�ӣ�֧�ւ�(g��)�Ի���ˎ,�� ���m(x��)���M(j��n)�c�ИI(y��)�����C(j��)�� EMAÿ��l(f��)��eCTD��(sh��)ʩ��(b��o)��,��������Ҋ�e(cu��)�`����ָ�ϡ��ИI(y��)(li��n)�ˣ���EFPIA��ͨ�^(gu��)������ӑ��(hu��)��O(ji��n)�ܙC(j��)��(g��u)�������g(sh��)ʹ�c(di��n),���Ƅ�(d��ng)��(bi��o)��(zh��n)��(y��u)��,���_��ʽAPI�ӿڵ��ƏV�����M(j��n)eCTD����朵Ļ������ԣ����ͼ��g(sh��)�i���L(f��ng)�U(xi��n),���㽭����ˎeCTD����(w��)�Ԓ��DMFע��(c��)���(b��o)���P(gu��n)���g(sh��)֧��,��

2015��l(f��)�����P(gu��n)��ˎƷ�t(y��)����е���u(p��ng)�����ƶȵ���Ҋ�������ˎ�O(ji��n)���Ŀ��(bi��o),����eCTD�{���(gu��)��ˎ�O(ji��n)��(sh��)�ֻ���(zh��n)��,��2017�꣬�Ї�(gu��)����ICH����(gu��)�H����ˎƷע��(c��)���g(sh��)�f(xi��)�{(di��o)��(hu��)��,���ɞ�ȫ��ڰ˂�(g��)�O(ji��n)�ܙC(j��)��(g��u)�ɆT,�������c��(gu��)�H��(bi��o)��(zh��n)��܉��2018��,����(gu��)��ˎ�O(ji��n)�֣�NMPA�����eCTD�ęn����ϵ�y(t��ng)�И�(bi��o),�����Ϻ������c��(gu��)LORENZ��������g(sh��)ƽ�_(t��i)����(bi��o)־�����g(sh��)���A(ch��)�O(sh��)ʩ�����,�� Ҏ(gu��)���ƶ��cԇ�c(di��n)�A�Σ�2019-2023�꣩ 2019-2020��,��CDE��ˎƷ���u(p��ng)���ģ��l(f��)����eCTD���g(sh��)Ҏ(gu��)�������(y��n)�C��(bi��o)��(zh��n)����������Ҋ�壬���M����݆��I(y��)�y(c��)ԇ,��2021��,��NMPA���_���W(xu��)ˎ1�5.1�������Ʒ1�������Ո(q��ng)�m��eCTD��2022�ꌍ(sh��)ʩ������(b��o)����eCTD��ʽ��,��2023��ȡ�����|(zh��)�Y���ύ,����eCTD��_�춨���A(ch��)�� ��(sh��)ʩ�c�U(ku��)չ�A�Σ�2024-2025�꣩ 2024��3�¸�������(b��o)���g(sh��)Ҫ��,��7����(d��ng)�W(w��ng)�j(lu��)��ݔԇ�c(di��n),��2025��1��27�գ�NMPA��eCTD�m�÷����U(ku��)������ˎ1-5��R��ԇ�(y��n)��������Ո(q��ng),��������Ʒ1-3�ȫ����,�����wˎ������ˎ���������ˎ,����(sh��)�F(xi��n)�c��(gu��)�H�������(b��o)ģʽͬ��,��
2020�걩�l(f��)��F(xi��n)DA�M(j��n)һ���Ƅ�(d��ng)��ӻ��M(j��n)��,���������S�h(yu��n)����Ӻ��º��R�r(sh��)�Ō����ָ�ʽҪ��,�����(y��n)�C��(bi��o)��(zh��n)����PDF�汾,������朽���Ч�ԣ���δ����,���@һ�r(sh��)�ڵČ�(sh��)�`��eCTD�ھo�������е��`�����ṩ�˰�����Ҳ�@��������Σ�C(j��)��(y��ng)��(du��)���ߵăr(ji��)ֵ,�� �M������(gu��)��δ����eCTD V4.0,�����似�g(sh��)���������_��֧���t(y��)����е�ͱ���Ʒ���(b��o)������(qi��ng)��(sh��)��(j��)�ɏ�(f��)����,����(y��u)�����u(p��ng)ϵ�y(t��ng)�c�˹����ܵļ���,�����⣬�^(q��)�K朼��g(sh��)����Ӻ��º͔�(sh��)��(j��)��Դ�еđ�(y��ng)��̽��,�����ܳɞ���һ�A������(j��)�����c(di��n)��ʿNDAע��(c��)���(b��o)���P(gu��n)���g(sh��)֧��,��
�ļ��������ڹ�����eCTD֧���ļ���Q��Replace�����h����Delete���Ȳ���,���������ļ�,�����磬���R���о������r(sh��)����Replace�������w�f�汾,�,������ύ��Baseline Submission���������a(b��)��vʷ���|(zh��)�Y�ϣ������ڷ��溯�����o(w��)��(n��i)��׃��,�� �R����(sh��)��(j��)�c�о���(bi��o)���ļ���STF����ģ�K4��5�е��о���(sh��)��(j��)��ͨ�^(gu��)STF��Study Tagging Files������,���_����(sh��)��(j��)�c�ęn�P(gu��n)(li��n)��FDAҪ��(sh��)��(j��)������SAS XPORT��ʽ��������ģ�K3-5,���҆�(g��)�ļ����^(gu��)4GB����,��2022��y(t��ng)Ӌ(j��)�@ʾ��58%��ANDA���о���(sh��)��(j��)���g(sh��)�ܽ^��(bi��o)��(zh��n)��TRC���e(cu��)�`���ܡ� ��Ӻ����c����Ҫ��FDA������356h,��1571����ʹ�Ô�(sh��)�ֺ���,��PDF�ļ���ֹ���ܻ��O(sh��)�þ����ơ���Ӻ��������21 CFR Part 11Ҏ(gu��)��,���_�������(y��n)�C,�����ɷ��J(r��n)�Ժ͔�(sh��)��(j��)�����ԡ� �������(w��)�cϵ�y(t��ng)��Q�������x���Ƽ���Ӌ(j��)�ύ��2000��eCTD��Ո(q��ng),������ɽ���40%�˹��e(cu��)�`��,���Ĵ�����eCTDע��(c��)��ԃ���P(gu��n)���g(sh��)֧�֡����Kԭ��ˎeCTD��ʲô
��ANDAע��(c��)���(b��o)���P(gu��n)���g(sh��)֧��,���㽭����ˎeCTD����(w��)�Ԓ
eCTD�ښW��ˎƷ�O(ji��n)���еĚvʷ�������W��eCTD�����ͨ�ü��g(sh��)�ęn���İl(f��)չʼ�ڌ�(du��)�R��ԇ�(y��n)��ˎƷ���u(p��ng)���̘�(bi��o)��(zh��n)��������,��2001�꣬�W�����롶�R��ԇ�(y��n)ָ���CTD������y(t��ng)һ�ķ��ɿ��,�������ɢ�ijɆT��(gu��)���(b��o)�C(j��)�ƌ�(d��o)��Ч�ʵ���,��2014�꣬�W��ͨ�^(gu��)���R��ԇ�(y��n)��Ҏ(gu��)����CTR, Regulation EU No. 536/2014��,��Ҫ��ͨ�^(gu��)CTISƽ�_(t��i)���R��ԇ�(y��n)��Ϣϵ�y(t��ng)�������ύ�R��ԇ�(y��n)��Ո(q��ng)��CTA��,�������Ƅ�(d��ng)eCTD������ӻ����(b��o)�Ĺ��ߡ��@һּ�ڽ�Q���y(t��ng)CTDģʽ���u(p��ng)�����L(zh��ng),������(gu��)�f(xi��)�{(di��o)�ɱ��ߵĆ�(w��n)�},����eCTD�Č�(sh��)ʩ�춨�˻��A(ch��)���㽭����ˎeCTD����(w��)�Ԓ
������̶��Ԓ��Ո(q��ng)?ji��n)څ^(q��)̖(h��o)�������"-"�� ��֙C(j��)̖(h��o)�������ˈ�(b��o)�r(ji��)�����M(f��i)���ն���֪ͨ
 �㽭��(gu��)�a(ch��n)eCTD���� �gӭ��(l��i)� �x���Ƽ�����(y��ng)
�㽭��(gu��)�a(ch��n)eCTD���� �gӭ��(l��i)� �x���Ƽ�����(y��ng)
���h
 �Ї�(gu��)�_(t��i)��ˎƷע��(c��)eCTD ����(w��)���� �x���Ƽ�����(y��ng)
�Ї�(gu��)�_(t��i)��ˎƷע��(c��)eCTD ����(w��)���� �x���Ƽ�����(y��ng)
���h
 �Ϻ���(gu��)�a(ch��n)eCTD�ļ�������� �gӭ��(l��i)� �x���Ƽ�����(y��ng)
�Ϻ���(gu��)�a(ch��n)eCTD�ļ�������� �gӭ��(l��i)� �x���Ƽ�����(y��ng)
���h
 �㽭INDeCTD���� �gӭ��ԃ �x���Ƽ�����(y��ng)
�㽭INDeCTD���� �gӭ��ԃ �x���Ƽ�����(y��ng)
���h
 ɽ�|ԭ��ˎeCTD����(w��)���Ŀɿ� ֵ����ه �x���Ƽ�����(y��ng)
ɽ�|ԭ��ˎeCTD����(w��)���Ŀɿ� ֵ����ه �x���Ƽ�����(y��ng)
���h