南京實(shí)時(shí)Real-time PCR應(yīng)用
聚合酶鏈?zhǔn)椒磻?yīng)的常見問題:靶序列或擴(kuò)增產(chǎn)物的交叉污染:這種污染有兩種原因:一是整個(gè)基因組或大片段的交叉污染,導(dǎo)致假陽(yáng)性,。這種假陽(yáng)性可用以下方法解決:操作時(shí)應(yīng)小心輕柔,,防止將靶序列吸入加樣內(nèi)或?yàn)R出離心管外。除酶及不能耐高溫的物質(zhì)外,,所有試劑或器材均應(yīng)高壓消毒,。所用離心管及樣進(jìn)頭等均應(yīng)一次性使用。必要時(shí),,在加標(biāo)本前,,反應(yīng)管和試劑用紫外線照射,以破壞存在的核酸,。二是空氣中的小片段核酸污染,,這些小片段比靶序列短,但有一定的同源性,??苫ハ嗥唇樱c引物互補(bǔ)后,,可擴(kuò)增出PCR產(chǎn)物,,而導(dǎo)致假陽(yáng)性的產(chǎn)生,可用巢式PCR方法來減輕或消除,。聚合酶鏈?zhǔn)椒磻?yīng)是80年代中期發(fā)展起來的體外核酸擴(kuò)增技術(shù),。南京實(shí)時(shí)Real-time PCR應(yīng)用
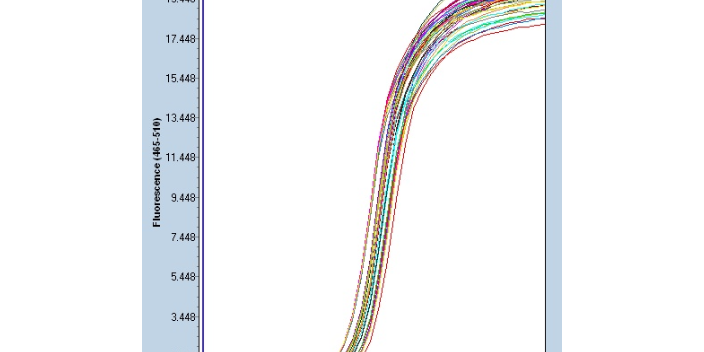
聚合酶鏈?zhǔn)椒磻?yīng)的試驗(yàn)污染:陽(yáng)性對(duì)照:在建立PCR反應(yīng)實(shí)驗(yàn)室及一般的檢驗(yàn)單位都應(yīng)設(shè)有PCR陽(yáng)性對(duì)照,它是PCR反應(yīng)是否成功,、產(chǎn)物條帶位置及大小是否合乎理論要求的一個(gè)重要的參考標(biāo)志,。陽(yáng)性對(duì)照要選擇擴(kuò)增度中等、重復(fù)性好,,經(jīng)各種鑒定是該產(chǎn)物的標(biāo)本,,如以重組質(zhì)粒為陽(yáng)性對(duì)照,其含量宜低不宜高(100個(gè)拷貝以下),。但陽(yáng)性對(duì)照尤其是重組質(zhì)粒及高濃度陽(yáng)性標(biāo)本,,其對(duì)檢測(cè)或擴(kuò)增樣品污染的可能性很大。因而當(dāng)某一PCR試劑經(jīng)自己使用穩(wěn)定,,檢驗(yàn)人員心中有數(shù)時(shí),,在以后的實(shí)驗(yàn)中可免設(shè)陽(yáng)性對(duì)照。陰性對(duì)照:每次PCR實(shí)驗(yàn)務(wù)必做陰性對(duì)照,。它包括:標(biāo)本對(duì)照:被檢的標(biāo)本是血清就用鑒定后的正常血清作對(duì)照,;被檢的標(biāo)本是組織細(xì)胞就用相應(yīng)的組織細(xì)胞作對(duì)照。試劑對(duì)照:在PCR試劑中不加模板DNA或RNA,進(jìn)行PCR擴(kuò)增,,以監(jiān)測(cè)試劑是否污染,。寧波微量Real-time PCR供應(yīng)商重疊延伸聚合酶鏈反應(yīng):一種用于將兩個(gè)或多個(gè)含有互補(bǔ)序列的DN段拼接在一起的基因工程技術(shù)。

聚合酶鏈反應(yīng)允許快速生產(chǎn)短DN段,,即使已知的引物序列不超過兩個(gè),。聚合酶鏈反應(yīng)的這種能力增強(qiáng)了許多方法,例如生成雜交 探針用于 Southern 或northern blot 雜交,。聚合酶鏈反應(yīng)為這些技術(shù)提供了大量的純DNA,,有時(shí)是單鏈的,甚至可以從非常少量的起始材料中進(jìn)行分析,。脫氧核糖核酸測(cè)序的任務(wù)也可以通過聚合酶鏈反應(yīng)來輔助完成,。已知的DN段可以很容易地從患有遺傳疾病突變的患者體內(nèi)產(chǎn)生。對(duì)擴(kuò)增技術(shù)的修飾可以從完全未知的基因組中提取片段,,或者可以產(chǎn)生感興趣區(qū)域的單鏈,。聚合酶鏈反應(yīng)有許多應(yīng)用于傳統(tǒng)的 DNA克隆。它可以從更大的基因組中提取片段以插入載體,,這可能只有少量可用,。使用一組“載體引物”,它還可以分析或提取已經(jīng)插入載體的片段,。聚合酶鏈反應(yīng)方案的一些改變可以產(chǎn)生突變(通用的或定點(diǎn)的)插入片段,。
聚合酶鏈?zhǔn)椒磻?yīng)的常見問題:Mg2+濃度:Mg2+離子濃度對(duì)PCR擴(kuò)增效率影響很大,濃度過高可降低PCR擴(kuò)增的特 異性,,濃度過低則影響PCR擴(kuò)增產(chǎn)量甚至使PCR擴(kuò)增失敗而不出擴(kuò)增條帶,。反應(yīng)體積的改變:通常進(jìn)行PCR擴(kuò)增采用的體積為20ul、30ul,、50ul,。或100ul,,應(yīng)用多 大體積進(jìn)行PCR擴(kuò)增,,是根據(jù)科研和臨床檢測(cè)不同目的而設(shè)定,在做小體積如20ul 后,,再做大體積時(shí),,一定要模索條件,否則容易失敗,。物理原因:變性對(duì)PCR擴(kuò)增來說相當(dāng)重要,,如變性溫度低,變性時(shí)間短,,極有可能出現(xiàn)假陰性,;退火溫度過低,,可致非特異性擴(kuò)增而降低特異性擴(kuò)增效率退火溫度過高影響引物與模板的結(jié)合而降低PCR擴(kuò)增效率。有時(shí)還有必要用標(biāo)準(zhǔn)的溫度計(jì),,檢測(cè)一下擴(kuò)增儀或水溶鍋內(nèi)的變性,、退火和延伸溫度,這也是PCR失敗的原因之一,。PCR的很大特點(diǎn)是能將微量的DNA大幅增加,。

聚合酶鏈?zhǔn)椒磻?yīng)是一種體外迅速擴(kuò)增DN段的技術(shù),它能以極少量的DNA為模版,,在幾小時(shí)內(nèi)復(fù)制出上百萬(wàn)份的DNA拷貝,。DNA的半保留復(fù)制是生物進(jìn)化和傳代的重要途徑。雙鏈DNA在多種酶的作用下可以變性解鏈成單鏈,,在DNA聚合酶與啟動(dòng)子的參與下,,根據(jù)堿基互補(bǔ)配對(duì)原則復(fù)制成同樣的兩分子挎貝。在實(shí)驗(yàn)中發(fā)現(xiàn),,DNA在高溫時(shí)由于兩條鏈之間的氫鍵被破壞也可以發(fā)生變性解鏈,,當(dāng)溫度降低后又可以復(fù)形成為雙鏈。因此,,通過溫度變化控制DNA的變性和復(fù)性,,并設(shè)計(jì)引物做啟動(dòng)子,加入DNA聚合酶,、dNTP就可以完成特定基因的體外復(fù)制,。但是,DNA聚合酶在高溫時(shí)會(huì)失活,,因此,,每次循環(huán)都得加入新的DNA聚合酶,不但操作煩瑣,,而且價(jià)格昂貴,,制約了PCR技術(shù)的應(yīng)用和發(fā)展。發(fā)現(xiàn)耐熱DNA聚合同酶--Taq酶對(duì)于PCR的應(yīng)用有里程碑的意義,,該酶可以耐受90℃以上的高溫而不失活,,不需要每個(gè)循環(huán)加酶,使PCR技術(shù)變得非常簡(jiǎn)捷,、同時(shí)也降低了成本,,PCR技術(shù)得以大量應(yīng)用,并逐步應(yīng)用于臨床,。PCR產(chǎn)物的電泳檢測(cè)時(shí)間一般為48h以內(nèi),,有些很好于當(dāng)日電泳檢測(cè),大于48h后帶型不規(guī)則甚至消失。蘇州分子生物學(xué)熒光定量PCR設(shè)計(jì)公司
聚合酶鏈?zhǔn)椒磻?yīng):DNA的半保留復(fù)制是生物進(jìn)化和傳代的重要途徑,。南京實(shí)時(shí)Real-time PCR應(yīng)用
聚合酶鏈?zhǔn)椒磻?yīng)的常見問題:出現(xiàn)非特異性擴(kuò)增帶:PCR擴(kuò)增后出現(xiàn)的條帶與預(yù)計(jì)的大小不一致,,或大或小,或者同時(shí)出現(xiàn)特異性擴(kuò)增帶 與非特異性擴(kuò)增帶,。非特異性條帶的出現(xiàn),,其原因:一是引物與靶序列不完全互補(bǔ)、或引物聚合形成二聚體,。二是Mg2+離子濃度過高、退火溫度過低,,及PCR循環(huán)次數(shù)過多有關(guān),。其次是酶的質(zhì)和量,往往一些來源的酶易出現(xiàn)非特異條帶而另一來源的酶則不出現(xiàn),,酶量過多有時(shí)也會(huì)出現(xiàn)非特異性擴(kuò)增,。其對(duì)策有:必要時(shí)重新設(shè)計(jì)引 物。減低酶量或調(diào)換另一來源的酶,。降低引物量,,適當(dāng)增加模板量,減少循環(huán)次數(shù),。適當(dāng)提高退火溫度或采用二溫度點(diǎn)法(93℃變性,,65℃左右退火與延伸)。南京實(shí)時(shí)Real-time PCR應(yīng)用
- 廈門光遺傳技術(shù)服務(wù)公司 2025-05-28
- 襄陽(yáng)組織芯片免疫熒光服務(wù)中心 2025-05-28
- 東莞化學(xué)遺傳技術(shù)用途 2025-05-28
- 上?;瘜W(xué)遺傳技術(shù)服務(wù)中心 2025-05-27
- 珠?;瘜W(xué)遺傳技術(shù)平臺(tái) 2025-05-27
- 徐州光遺傳技術(shù)哪家靠譜 2025-05-27
- 佛山光遺傳技術(shù)用途 2025-05-27
- 南通光遺傳技術(shù)哪家專業(yè) 2025-05-27
- 蚌埠光遺傳膜片鉗技術(shù)哪家專業(yè) 2025-05-27
- 淮南組織芯片免疫組化服務(wù)中心 2025-05-27
- 3D打印可調(diào)脊柱側(cè)彎矯形器 2025-05-28
- 安徽化學(xué)發(fā)光凍干試劑功效 2025-05-28
- 蘇州一次性血液過濾器ODM流程 2025-05-28
- 江蘇比較好的白夫康表皮抑菌膏出廠價(jià) 2025-05-28
- 廣西一維運(yùn)動(dòng)混合機(jī)哪里有賣的 2025-05-28
- 山西質(zhì)量中鹽核酸酶聯(lián)系方式 2025-05-28
- 濟(jì)寧高分子生物涂層應(yīng)用 2025-05-28
- 雨花臺(tái)區(qū)環(huán)境監(jiān)測(cè)儀器歡迎選購(gòu) 2025-05-28
- 楊浦區(qū)靠譜的醫(yī)療管理服務(wù)主題 2025-05-28
- 浙江患者信賴的自動(dòng)糞菌分離儀運(yùn)行時(shí)間 2025-05-28