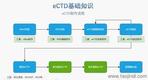
���K�Ї�eCTD��(b��o)�r(ji��) �\�Ż��� �x���Ƽ�����(y��ng)
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-23
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-23
�W��eCTD���f��;���c���g(sh��)Ҫ�� ��ͬ���u����?q��)��?y��ng)��ͬ�f�����������г���CP��ͨ�^EMA��eSubmission Gateway��Web Client�ύ����ɢ����DCP���ͻ��J(r��n)����MRP���t��ʹ�ÚW��ͨ���ύ�T����CESP��,���ļ��Y(ji��)��(g��u)���(y��n)����ѭģ�K��Ҫ��,������CEP��Ո�����ģ�K1�������ļ�����ģ�K2���|(zh��)����������ģ�K3�����g(sh��)�ęn��,����XML�����ļ�횷���EDQM���ض�����Ҏ(gu��)�t,�����⣬����PDF�ļ���o�ܴa���o(h��),����ȫ�ęz��,����Ƕ��Ӽ�������֧�ֿ��ٌ�醡� CEP��Ո��eCTD�f�������� CEP������2018������(qi��ng)�Ʋ���eCTD��ʽ,�����c(di��n)�u��ԭ��ˎ�Ƿ���ϚW��ˎ���(bi��o)��(zh��n),����ģ�K1�����EDQM��Ո�������v��׃���f����,��ģ�K2��ʹ��EDQM�ṩ���|(zh��)������ģ��,��ģ�K3�t��CTD��ʽ�M��3.2.S�¹�(ji��)��(n��i)�ݡ�CEP�cASMF���������|(zh��)���ļ�������Ҫ�^(q��)�e�����ԣ�CEP�o���P(gu��n)(li��n)�����S��,���Ҍ��u��EDQM���,������eCTD�(y��n)�C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧�֡����K�Ї�eCTD��(b��o)�r(ji��)

������2003��ɞ�ȫ�������eCTD�����ͨ�ü��g(sh��)�ęn���ć���֮һ,������CDER��CBER��������ύƽ�_ԇ�c(di��n),��2008����eCTD��ʽ�ɞ�ˎ��Ո��NDA����������Ʒ�S����Ո��BLA���Ę�(bi��o)��(zh��n)��ʽ,������2012��ͨ�^��ˎ���(b��o)�߸��M(f��i)��������PDUFA���M(j��n)һ����(qi��ng)���䷨�ɵ�λ,����2017�꣬F(xi��n)DA��(qi��ng)��Ҫ������ˎ��Ո��NDA��,������ˎ��Ո��ANDA����ˎ�����ļ���DMF����횲���eCTD��ʽ�ύ,����(bi��o)־����Ŀ��x����(qi��ng)�Ƶ��D(zhu��n)�͡��@һ�M(j��n)����2018��U(ku��)չ���R��ԇ�(y��n)��Ո��IND��,���K��(sh��)�F(xi��n)ȫ���ˎƷע�Ե���ӻ����w�ؑc���W(xu��)ˎƷeCTD����ע��������̖��Ո���P(gu��n)���g(sh��)֧��,��
eCTD�ķ�Ҏ(gu��)����c���g(sh��)Ҏ(gu��)�����W��eCTD�ķ�Ҏ(gu��)�Ӽ�����ָ�ϣ�Guidelines��,��ָ�Directive���ͷ�Ҏ(gu��)��Regulation��������,����Ҏ(gu��)����CTR������ֱ�ӷ���Ч��,����ָ�ϣ���ICH eCTDҎ(gu��)�����t�鼼�g(sh��)�����ṩ������eCTD�ĽY(ji��)��(g��u)����ϚW��ģ�K1Ҏ(gu��)����DTD 3.0+��,�����������ļ���ģ�K1��,���|(zh��)����(sh��)��(j��)��ģ�K3�����R���о���(b��o)�棨ģ�K5���ȃ�(n��i)��,����ͨ�^XML�ļ���(sh��)�F(xi��n)��(sh��)��(j��)��(li��n),�����磬CEP���W��ˎ���m�����C������eCTD���(b��o)��Ϊ�(d��)��(g��u)���ŷ⣨Envelope����ģ�K1,����ָ����(bi��o)�R����UUID���Դ_�����g(sh��)��Ҏ(gu��)��,��
eCTD�Č�(sh��)ʩ��O(ji��n)�ܙC(j��)��(g��u)����I(y��)�����˶��ؙC(j��)������ӻ����(b��o)�Y���܉�O��ؼ��ٌ��uЧ��,���p���˞��Д��e(cu��)�`�͔�(sh��)��(j��)��������r,���Ķ���ߌ��u�Ĝ�(zh��n)�_�Ժ��ٶȡ�ͬ�r(sh��),��eCTD�����Ĕ�(sh��)��(j��)��(bi��o)��(zh��n)���C(j��)��ʹ��ȫ��O(ji��n)�ܙC(j��)��(g��u)���Y�σ�(n��i)�ݺ���Ӹ�ʽ���Խy(t��ng)һ,���������ڲ�ͬ�O(ji��n)�ܙC(j��)��(g��u)֮�g�M(j��n)�Д�(sh��)��(j��)��ݔ�������@��������ȫ��O(ji��n)��Ч�ʺ��ИI(y��)�аl(f��)Ч�ʾ�����Ҫ���x,�� ����,��eCTD�Č�(sh��)ʩ߀���M(j��n)�ˇ��H��������(g��u)����ȫ��O(ji��n)�ܵĵӴ�(sh��)��(j��)���A(ch��),��������I(y��)����,��eCTD�ṩ��һ��(g��)Ҏ(gu��)�������аl(f��)���ģ�壬�����ڽ����c�O(ji��n)�ܙC(j��)��(g��u)��ͨ�ijɱ�,��������(b��o)Ч��,���e�nj��ڇ���(n��i)�����\�g(sh��)��I(y��)���ԣ�eCTD�Č�(sh��)ʩ���Ǿ�����Ҫ���x,���������@Щ��I(y��)���õ�������H�Ј�,��Ȼ������С��I(y��)�������@Щ�C(j��)����ͬ�r(sh��),��Ҳ���R�����g(sh��)�ͳɱ�����,��eCTD�Č�(sh��)ʩ��Ҫ���T�ĈF(tu��n)�(du��)�M(j��n)��ϵ�y(t��ng)�S�o(h��)���_�l(f��)���@������С��I(y��)���f��һ�P��С���_֧,��ͬ�r(sh��),����(sh��)��(j��)��ȫ���}Ҳ����I(y��)�P(gu��n)ע�Ľ��c(di��n)�� �˴�CDE�U(ku��)��eCTD��(sh��)ʩ�������ИI(y��)������һ��(g��)�e�O���L(f��ng)���(bi��o),�����ڃ�(n��i),����I(y��)���R������(zh��n)�����m��(y��ng)����Ҫ��ļ��g(sh��)Ҏ(gu��)��������ļ��|(zh��)��,����eCTD����ϵ�y(t��ng)��ĥ���Լ��M(j��n)��eCTD֪�R�Ŀ�����Ӗ(x��n)�ȡ� �Ĵ�����eCTD���(b��o)ܛ�����P(gu��n)���g(sh��)֧��,��
����ˎ�������ˎ��ɼ����c��ؓ(f��)��(d��n)�Ե�һ�ˎ��,��2012����ǰ��ע�Ԍ��u�Dz���ȡ�κ��M(f��i)�õ�,������(d��ng)�r(sh��)����ˎ��Ո�e����(y��n)��,�������(b��o)���@����Ҫ3~5��ĕr(sh��)�g�� ����������2012���C���˷���ˎʹ�����M(f��i)����������Generic Drug User Fee Amendments, GDUFA��,��ԓ����Ҫ����ˎ�ИI(y��)֧��һ�����Ñ��M(f��i)��,�����a(b��)�����ˎ��Ո�Č��u�Լ��F(xi��n)���z����M(f��i)�ã��p�ٷ���ˎ��Ո�e��,���s�̌��u�r(sh��)�g,�����ӻ����L(f��ng)�U(xi��n)�ĬF(xi��n)���z��ȣ���Ŀ���Ǽӿ칫���@�ð�ȫ��Ч�ķ���ˎ,���������ИI(y��)�ɱ�,�� GDUFA���ÿ�������ڙ�(qu��n)һ�Σ���2017�����GDUFA II��,����2022�����GDUFA III��,�� Ŀǰ���M(f��i)�N֞������ķN��ANDA���u�M(f��i)��DMF���u�M(f��i),���ڌ��u�r(sh��)һ�����U�{,���(xi��ng)Ŀ�M(f��i)��Program fee)���O(sh��)ʩ�M(f��i)��Facility fee��,�������к�ÿ���U�{һ��,������eCTDע����ԃ���P(gu��n)���g(sh��)֧�֡�����eCTD�ԃr(ji��)�ȸ�
�Ĵ�������eCTD���(b��o)���P(gu��n)���g(sh��)֧��,�����K�Ї�eCTD��(b��o)�r(ji��)
�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ��,������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)��,����J(r��n)����MRP����,�������ɆT����RMS�����u����(b��o)���豻�����ɆT���J(r��n)�ɣ������F(xi��n)��������CMDh�f(xi��)�{(di��o)���ύEMA�ٲ�,���@�N���Ӽ����u�C(j��)��Ҫ����Ո�����ļ���(zh��n)���A�μ����]�^(q��)�������,��������m(x��)�������`�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y(ji��)��(g��u),��֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е�����(b��o),��������(qi��ng)�������ڹ������ܡ��W��Ӌ(j��)��ͨ�^2024��ԇ�c(di��n)���^����4.0�����ƽ���ļ��M����ʽ�����p���؏�(f��)�ύ���������uЧ��,��Ȼ��,����(sh��)ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m��(y��ng)�Ԇ��}�����K�Ї�eCTD��(b��o)�r(ji��)
������̶��Ԓ,��Ո?ji��n)څ^(q��)̖�������"-"�� ��֙C(j��)̖�������ˈ�(b��o)�r(ji��)�����M(f��i)���ն���֪ͨ