-
航瑞智能助力維尚家具打造自動倉儲系統(tǒng),,實現(xiàn)成品物流智能化升級
-
航瑞智能:準確把握倉儲痛點,,打造多樣化智能倉儲方案
-
高度集成化自動化立體倉庫:開啟高效物流新時代_航瑞智能
-
探秘倉儲物流中心:輸送機與RGV打造高效智能物流體系
-
共享裝備攜手航瑞智能打造砂芯智能倉儲,實現(xiàn)倉儲物流智能化升級
-
桁架機械手與輸送機:打造高效智能流水線
-
?采用WMS倉庫管理系統(tǒng)能夠給企業(yè)帶來哪些好處,?
-
?航瑞智能:精細把握倉儲痛點,,打造多樣化智能倉儲方案
-
往復式提升機:垂直輸送系統(tǒng)的智能化解決方案
-
航瑞智能:準確把握倉儲痛點,打造多樣化智能倉儲方案
江蘇國內(nèi)注冊eCTD服務商
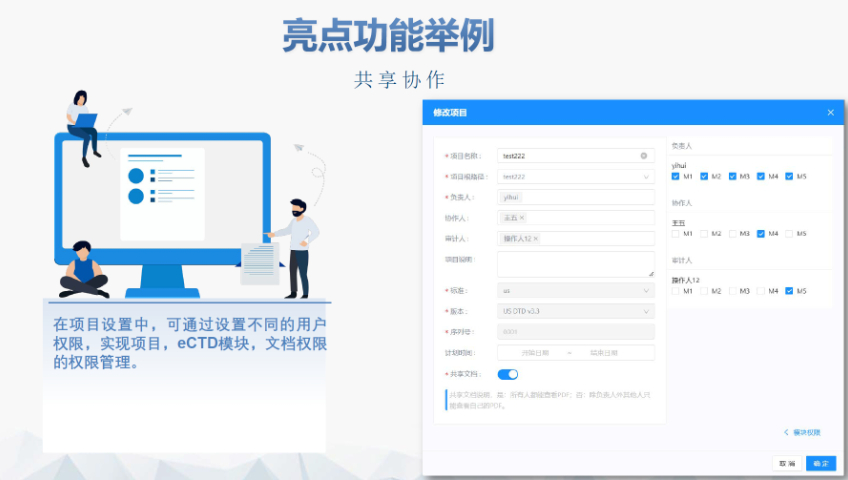
申報流程與要求 ?資料準備 ?內(nèi)容要求:包括產(chǎn)品描述,、生產(chǎn)工藝(原材料來源,、設備參數(shù)等)、質量控制標準(SOP,、穩(wěn)定性數(shù)據(jù)),、安全性與毒性研究等。 ?格式規(guī)范: 采用CTD(通用技術文件)格式,,按模塊分章節(jié)(如模塊3為CMC數(shù)據(jù)),。 電子提交需符合eCTD標準(文件小于10GB通過ESG系統(tǒng)提交,超過可選用CD-ROM),。 ?提交與注冊 ?預分配DMF號:需在提交前申請,,確保文件與編號綁定。 ?授權書(LOA)?:需向引用DMF的制劑廠商提供授權信,,明確可查閱的章節(jié),。 ?費用:Ⅱ類原料藥DMF需繳納年費(2024年約9,468美元)。 ?FDA審核流程 ?行政審評:2-3周內(nèi)確認文件完整性,。 ?完整性審評(CA)?:針對Ⅱ類DMF,,約60天。 ?技術審評:在DMF被制劑申請(如ANDA,、NDA)引用時啟動,,周期60-180天。 ?結果反饋:FDA可能要求補充數(shù)據(jù),,但DMF本身無“批準”狀態(tài),,通過后可能收到“無進一步意見函”(No Further Comment Letter)。中DMF注冊申報相關技術支持,。江蘇國內(nèi)注冊eCTD服務商
美國eCTD驗證采用三級分類:“錯誤”(必須修正),、“警告”(建議修正),、“提示信息”(參考)。例如,,PDF文件版本不符或加密保護屬于“錯誤”,,而書簽路徑非相對性則可能列為“警告”,。驗證失敗將直接導致退審,,企業(yè)需通過LORENZ Validator等工具預檢,確保提交前合規(guī),。 ?技術驗證點 驗證涵蓋XML結構合規(guī)性,、文件命名規(guī)則、生命周期管理(如序列號連續(xù)性)及PDF屬性(如字體嵌入,、可搜索性),。臨床試驗數(shù)據(jù)需額外滿足CDISC標準,包括SDTM和ADaM數(shù)據(jù)集的結構驗證吳江區(qū)中國eCTD哪家好美國eCTD注冊外包相關技術支持,。
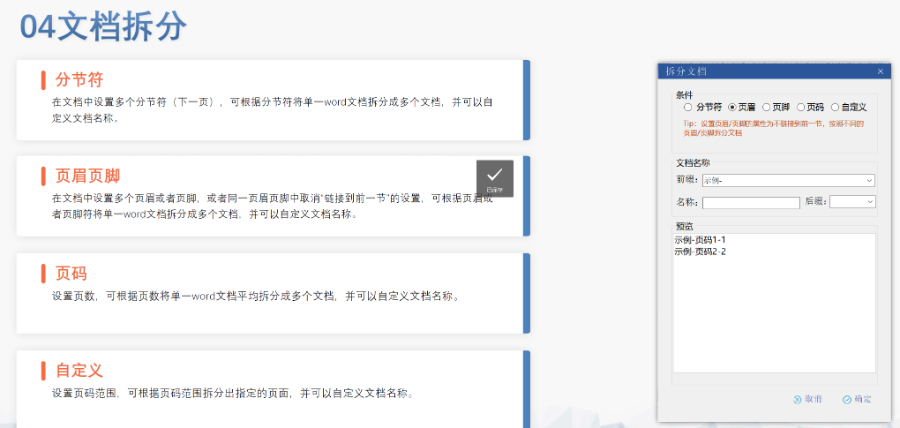
內(nèi)容與格式檢查Word預處理:需檢查拼寫,、縮略語、單位格式(如),,設置多級列表自動編號(如),,統(tǒng)一字體(宋體/TimesNewRoman)和段落格式。重復內(nèi)容處理:相同劑型不同規(guī)格可共用模塊3,,但需區(qū)分包裝系統(tǒng)(如,、)。外文資料:中文在前,、原文在后,,參考文獻需中英文對照并建立跨網(wǎng)頁鏈接。使用符合ICH標準的eCTD編輯器自動生成XML骨架和MD5校驗值,,拖拽PDF文件構建結構樹,。序列管理:序列號從0000開始遞增,每次提交需更新序列,,生命周期狀態(tài)(New/Replace/Delete)需在XML中明確標注,。驗證與遞交:確保無驗證錯誤(如書簽缺失、超鏈接斷鏈),,通過ESG等電子通道傳輸,,光盤封面需包含申請?zhí)柡托蛄刑枴H芷诠芾戆姹荆和ㄟ^軟件實現(xiàn)網(wǎng)頁簽入/簽出,、審批流程,,支持歷史版本追溯。變更管理:增補(Append)和替換(Replace)需關聯(lián)原始序列,,刪除(Delete)需徹底移除無效文件,。
審評效率與時間線優(yōu)化 eCTD的標準化縮短了審評周期:集中程序平均審評時間從18個月降至12個月,互認程序可在90天內(nèi)完成成員國意見協(xié)調(diào)。自動化驗證工具減少了格式錯誤導致的退審率,,但復雜藥學數(shù)據(jù)的科學審評仍需較長時間,。申請人可通過預提交會議(Pre-submission meeting)提前溝通技術細節(jié),規(guī)避潛在延誤,。 區(qū)域協(xié)作與全球互認 歐盟通過互認程序與澳大利亞,、加拿大等國實現(xiàn)eCTD數(shù)據(jù)共享,CEP證書在40余個非歐盟國家有效,。然而,,模塊一區(qū)域信息的差異性仍要求申請人定制化調(diào)整,例如亞洲國家可能要求附加穩(wěn)定性研究數(shù)據(jù),。ICH的協(xié)調(diào)作用有助于減少重復提交,,但完全全球化仍需解決法規(guī)和技術壁壘。 技術工具與行業(yè)生態(tài) 主流eCTD編輯軟件(如Lorenz,、Extedo)支持歐盟區(qū)域模板的自動化生成,,并與驗證工具集成實現(xiàn)一鍵校驗。云平臺解決方案逐漸普及,,支持多國團隊協(xié)同編輯和實時版本控制,。然而,軟件采購和維護成本較高,,中小企業(yè)常選擇外包給專業(yè)服務商完成遞交,。歐盟eCTD驗證標準相關技術支持。
設施費動態(tài)調(diào)整 API工廠和制劑工廠年費分別約6.8萬和14.5萬美元(2025財年),,CMO工廠費用為制劑費的24%,。國外工廠需額外支付1.5萬美元跨境檢查費。 ?繳費時限與懲罰 費用需在財年首日(10月1日)起20天內(nèi)繳納,,逾期將列入拖欠名單并暫停ANDA受理,,涉事藥品視為冒牌產(chǎn)品。 ?豁免與特殊情形 PET藥物,、非商業(yè)產(chǎn)品及停產(chǎn)超一年的工廠可豁免繳費,。已繳費工廠若年度內(nèi)無生產(chǎn)活動,仍需繳納費用,。 ?行業(yè)影響與策略 費用上漲推動企業(yè)優(yōu)化申報策略,,例如集中ANDA提交周期、采用CMO外包降低設施費,,并通過預認證(如DMF完整性評估)減少重復支出,。 澳大利亞DMF注冊申報相關技術支持。山東中國eCTD軟件
瑞士eCTD驗證標準相關技術支持,。江蘇國內(nèi)注冊eCTD服務商
美國藥物主文件(Drug Master File, DMF)是向FDA提交的機密技術文件,,用于支持藥品生產(chǎn),、質量控制及合規(guī)性審查。以下為申報的要點和流程總結: DMF概述與類型 ?定義與作用 DMF是藥品生產(chǎn)全過程的詳細檔案,,包含原料藥,、輔料、包裝材料等的生產(chǎn)設施,、工藝,、質量控制等信息,供制劑廠商引用以支持其注冊申請,。其意義在于保護企業(yè)機密的同時,,滿足FDA對供應鏈透明度的要求,。 ?DMF類型 ?Ⅱ類:原料藥,、中間體及制劑(如微生物外泌體、細胞株等生物制品均屬此類),。 ?Ⅲ類:包裝材料,。 ?Ⅳ類:輔料、著色劑等添加劑,。 ?Ⅴ類:非臨床/臨床數(shù)據(jù)等特殊信息(需FDA預先批準),。 注:Ⅰ型(生產(chǎn)設施與人員)已于2000年停用。江蘇國內(nèi)注冊eCTD服務商
- 高新區(qū)中國eCTD歡迎選購 2025-05-14
- 靜安區(qū)生物制品eCTD使用 2025-05-14
- 蕪湖新藥eCTD是什么 2025-05-14
- 吳江區(qū)賦悅科技eCTD供應商 2025-05-14
- 南京生物制品eCTD注冊系統(tǒng) 2025-05-14
- 上?;瘜W藥品eCTD格式 2025-05-09
- 南京電子申報eCTD哪個品牌好 2025-05-09
- 太倉NDAeCTD服務價格 2025-05-09
- 浦東新區(qū)原料藥eCTD文件如何制作 2025-04-26
- 南京新藥eCTD找哪家 2025-04-26
- 寧波儲能線束定制 2025-06-04
- ??谖臅浵駧?shù)據(jù)轉換 2025-06-04
- 惠州硬盤報價 2025-06-04
- 安防監(jiān)控識別價格 2025-06-04
- 楊浦區(qū)品牌搜索引擎代運營 2025-06-04
- 成都醫(yī)療內(nèi)窺鏡攝像頭模組聯(lián)系方式 2025-06-04
- 共享無線充開發(fā) 2025-06-04
- 沈陽直流充電槍測試儀廠商 2025-06-04
- 自動化銷售云T云銷售公司 2025-06-04
- 衡陽定制商用電腦設備租賃方案 2025-06-04