-
航瑞智能助力維尚家具打造自動倉儲系統(tǒng),,實現(xiàn)成品物流智能化升級
-
航瑞智能:準確把握倉儲痛點,,打造多樣化智能倉儲方案
-
高度集成化自動化立體倉庫:開啟高效物流新時代_航瑞智能
-
探秘倉儲物流中心:輸送機與RGV打造高效智能物流體系
-
共享裝備攜手航瑞智能打造砂芯智能倉儲,實現(xiàn)倉儲物流智能化升級
-
桁架機械手與輸送機:打造高效智能流水線
-
?采用WMS倉庫管理系統(tǒng)能夠給企業(yè)帶來哪些好處,?
-
?航瑞智能:精細把握倉儲痛點,,打造多樣化智能倉儲方案
-
往復式提升機:垂直輸送系統(tǒng)的智能化解決方案
-
航瑞智能:準確把握倉儲痛點,,打造多樣化智能倉儲方案
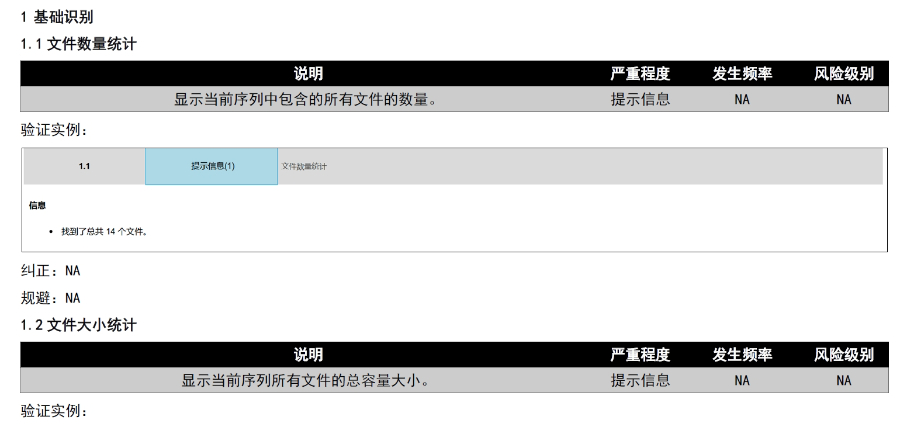
山東國產(chǎn)eCTD發(fā)布軟件
2020年暴發(fā)后,F(xiàn)DA進一步推動電子化進程,,例如允許遠程電子簽章和臨時放寬部分格式要求,,但驗證標準(如PDF版本、書簽鏈接有效性)并未降低,。這一時期的實踐為eCTD在緊急審批中的靈活性提供了案例,,也凸顯了其作為危機應對工具的價值。 盡管美國尚未部署eCTD V4.0,,但其技術方向已明確:支持醫(yī)療器械和保健品申報,、增強數(shù)據(jù)可復用性、優(yōu)化審評系統(tǒng)與人工智能的集成,。此外,,區(qū)塊鏈技術在電子簽章和數(shù)據(jù)溯源中的應用探索,可能成為下一階段升級的重點瑞士DMF注冊申報相關技術支持,。山東國產(chǎn)eCTD發(fā)布軟件
仿制藥作為提高藥物可及性與可負擔性的一類藥物,,2012年以前,注冊審評是不收取任何費用的,,但當時仿制藥申請積壓嚴重,,從申報到獲批需要3~5年的時間。 美國國會于2012年頒布了仿制藥使用者費用修正案(Generic Drug User Fee Amendments, GDUFA),,該法律要求制藥行業(yè)支付一定的用戶費用,,以補充仿制藥申請的審評以及現(xiàn)場檢查的費用,減少仿制藥申請積壓,,縮短審評時間,,增加基于風險的現(xiàn)場檢查等,其目的是加快公眾獲得安全有效的仿制藥,并降低行業(yè)成本,。 GDUFA必須每五年重授權一次,,于2017年更(GDUFA II),,于2022年更(GDUFA III),; 目前收費種類分為以下四種:ANDA審評費、DMF審評費,,在審評時一次性繳納,;項目費(Program fee)、設施費(Facility fee),,是上市后每年繳納一次,。寧波國產(chǎn)eCTD報價澳大利亞DMF注冊申報相關技術支持。
美國于2003年成為全球早采用eCTD(電子通用技術文檔)的國家之一,,初由CDER和CBER作為電子提交平臺試點,。2008年起,eCTD正式成為藥申請(NDA)和生物制品許可申請(BLA)的標準格式,,并在2012年通過《藥申報者付費法案》(PDUFA)進一步強化其法律地位,。至2017年,F(xiàn)DA強制要求所有藥申請(NDA),、簡略藥申請(ANDA)及藥物主文件(DMF)必須采用eCTD格式提交,,標志著其從可選到強制的轉型。這一進程在2018年擴展至臨床試驗申請(IND),,終實現(xiàn)全類型藥品注冊的電子化覆蓋
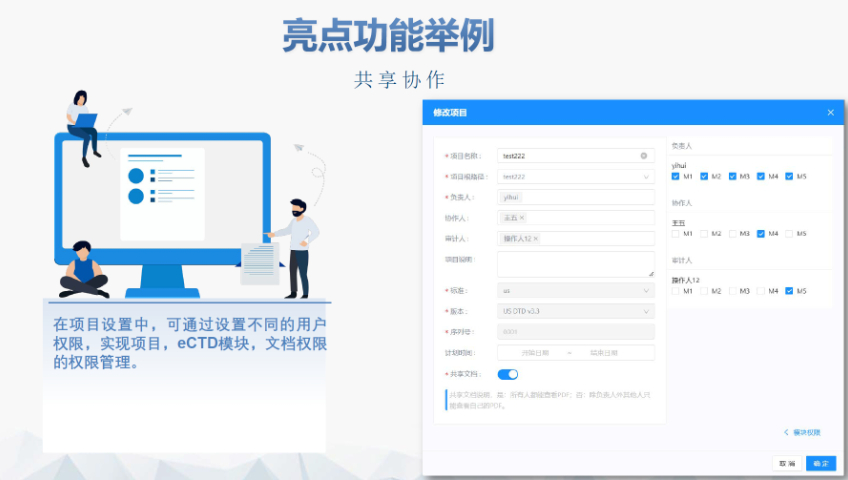
DMF維護與合規(guī) ?年度更 即使無變更,,每年需提交聲明;重大工藝/設施變更需及時通知客戶并更文件,。 ?現(xiàn)場檢查 原料藥企業(yè)需通過FDA現(xiàn)場檢查,,驗證是否符合ICH Q7 GMP標準,并與DMF內(nèi)容一致,。 ?轉讓與關閉 ?轉讓:需書面通知FDA并提供持有者信息,。 ?關閉:未提交年度報告或持有人主動申請,需說明原因并通知所有授權方,。 關鍵注意事項 ?數(shù)據(jù)質量:所有資料需準確,、完整,減少審核延遲風險,。 ?合規(guī)性:遵循FDA指南(如21 CFR Part 207)及USP標準(如培養(yǎng)基物料來源級別),。 ?溝通機制:建議通過專業(yè)機構(如瑞歐佰藥)協(xié)助,定期提交周報并制定計劃表以提高效率,。 常見問題解答 ?生物制品分類:培養(yǎng)基,、外泌體等均屬Ⅱ類DMF。 ?質量標準:參考USP及同行標準,需提供分析方法驗證及雜質對比研究,。 ?周期估算:資料準備約5-50個工作日,,總周期受缺陷回復影響。eCTD驗證實踐手冊相關技術支持,。
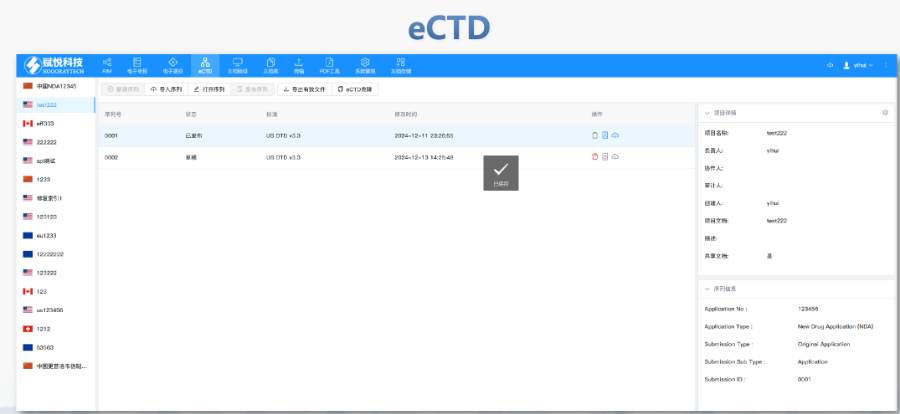
歐洲藥品管理局:集中審評程序由歐洲藥品管理局(European Medicines Agency, EMA)負責協(xié)調,。 人用藥品委員會:人用藥品委員會(Committee for Medicinal Products for Human Use, CHMP)負責提供科學意見。 歐盟委員會:CHMP的意見隨后被提交給歐盟委員會(European Commission, EC),,由歐盟委員會做出是否授權的終決定,。這個決定在整個歐盟都是具有法律約束力的。 審批過程: 申請人向EMA提交申請,,包括eCTD(電子通用技術文檔)格式的藥品注冊文檔,。 EMA的CHMP分配一個科學評估團隊(Rapporteur和Co-Rapporteur),負責初步評估,。 CHMP基于評估團隊的報告提供科學意見,。 歐盟委員會根據(jù)CHMP的意見做出終決定,批準或拒絕藥品上市,。 授權范圍 如果藥品獲得批準,,將獲得在整個歐盟、冰島,、列支敦士登和挪威有效的上市許可(Central Marketing Authorisation, CMA),。澳大利亞IND注冊申報相關技術支持。寧波仿制藥eCTD格式
加拿大NDA注冊申報相關技術支持,。山東國產(chǎn)eCTD發(fā)布軟件
技術壁壘與興市場挑戰(zhàn) 非洲和東南亞國家逐步采納eCTD,,但其IT基礎設施薄弱導致實施進度滯后。歐盟通過“eCTD全球化倡議”提供技術援助,,幫助興市場建立驗證體系和培訓中心,。跨國藥企需針對不同區(qū)域定制遞交策略,,例如在模塊1附加本地穩(wěn)定性數(shù)據(jù),。 監(jiān)管科學與創(chuàng)激勵 eCTD支持真實世界證據(jù)(RWE)和適應性臨床試驗設計的整合,加速創(chuàng)藥上市,。EMA的PRIME計劃為突破性療法提供eCTD快速通道,,允許分階段提交模塊數(shù)據(jù)。孤兒藥和兒科藥的eCTD序列可享受費用減免和優(yōu)先審評,。 供應鏈安全與審計追蹤 eCTD的XML主干文件記錄所有提交版本,,支持供應鏈問題的追溯分析。原料藥CEP持有者需及時更變更信息,,確保下游制劑廠商獲取數(shù)據(jù),。區(qū)塊鏈技術試點用于追蹤eCTD數(shù)據(jù)流,,防止篡改和未授權訪問。 文化差異與實施障礙 部分南歐國家偏好傳統(tǒng)紙質流程,,導致eCTD推廣阻力較大,。EMA通過多語種培訓材料和區(qū)域協(xié)調員制度促進文化適應。行業(yè)需調整管理思維,,將eCTD從“合規(guī)負擔”轉化為“競爭優(yōu)勢”,。山東國產(chǎn)eCTD發(fā)布軟件
- 高新區(qū)中國eCTD歡迎選購 2025-05-14
- 靜安區(qū)生物制品eCTD使用 2025-05-14
- 蕪湖新藥eCTD是什么 2025-05-14
- 吳江區(qū)賦悅科技eCTD供應商 2025-05-14
- 南京生物制品eCTD注冊系統(tǒng) 2025-05-14
- 上海化學藥品eCTD格式 2025-05-09
- 南京電子申報eCTD哪個品牌好 2025-05-09
- 太倉NDAeCTD服務價格 2025-05-09
- 浦東新區(qū)原料藥eCTD文件如何制作 2025-04-26
- 南京新藥eCTD找哪家 2025-04-26
- 上海什么是游戲開發(fā)技術指導 2025-06-04
- 湖南高級DC380證卡打印機制造廠家 2025-06-04
- 江西電商蜂蜜能吃嗎 2025-06-04
- 惠州校園智慧食堂管理系統(tǒng) 2025-06-04
- 安徽什么是電商平臺代運營 2025-06-04
- 西南農(nóng)機軟件廠商 2025-06-04
- 寧波智能化游戲開發(fā)產(chǎn)品 2025-06-04
- 諸暨墨倉式復印機租賃市場價 2025-06-04
- 零配件系統(tǒng)哪些好用 2025-06-04
- 啟東社交平臺代運營圖片 2025-06-04