�㽭����ˎeCTDע��ϵ�y(t��ng) �\�Ž�(j��ng)�I �x���Ƽ�����(y��ng)
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-23

�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-23
�W��eCTD���f��;���c���g(sh��)Ҫ�� ��ͬ���u����?q��)��?y��ng)��ͬ�f�����������г���CP��ͨ�^EMA��eSubmission Gateway��Web Client�ύ,����ɢ����DCP���ͻ��J(r��n)����MRP���t��ʹ�ÚW��ͨ���ύ�T����CESP�����ļ��Y(ji��)��(g��u)���(y��n)����ѭģ�K��Ҫ��,������CEP��Ո�����ģ�K1�������ļ���,��ģ�K2���|(zh��)����������ģ�K3�����g(sh��)�ęn������XML�����ļ�횷���EDQM���ض�����Ҏ(gu��)�t,������,������PDF�ļ���o�ܴa���o(h��)����ȫ�ęz��,����Ƕ��Ӽ�������֧�ֿ��ٌ��,�� CEP��Ո��eCTD�f�������� CEP������2018������(qi��ng)�Ʋ���eCTD��ʽ�����c(di��n)�u��ԭ��ˎ�Ƿ���ϚW��ˎ���(bi��o)��(zh��n),����ģ�K1�����EDQM��Ո��,�����v��׃���f������ģ�K2��ʹ��EDQM�ṩ���|(zh��)������ģ��,��ģ�K3�t��CTD��ʽ�M��3.2.S�¹�(ji��)��(n��i)��,��CEP�cASMF���������|(zh��)���ļ�������Ҫ�^(q��)�e�����ԣ�CEP�o���P(gu��n)(li��n)�����S�ɣ��Ҍ��u��EDQM���,���W��DMFע�����(b��o)���P(gu��n)���g(sh��)֧��,���㽭����ˎeCTDע��ϵ�y(t��ng)
�x��eCTDϵ�y(t��ng) �ļ��(y��n)�C�c�ޏ�(f��) ֧���Ԅ��(y��n)�C�ļ���ʽ����PDF���ԡ����wǶ��,����朽������Եȣ�,����һ�I�ޏ�(f��)�����Ϸ�Ҏ(gu��)Ҫ����ļ�������,��ϵ�y(t��ng)���Ԅәz��XML�����ļ��ĽY(ji��)��(g��u)��Ҏ(gu��)��,���_�������Ї�������,���W�˵ȵ^(q��)��eCTD��Ҏ(gu��)��(bi��o)��(zh��n),�� eCTD�M�b�c�l(f��)�� ���Ԅ����ɷ���CTD�Y(ji��)��(g��u)������ęn��������XML�����ļ�,���ļ��A����Ҏ(gu��)��������̖����������Ո?zh��)?����̖�ļ��A�Ԅ����ɣ�,����֧�ֳ�朽Ӻ͕�����������(chu��ng)��������,�������ύ������̖��0000,�����m(x��)ÿ���ύ�Ԅ��f��,�� �������ڹ��� ֧���ļ�ȫ�������ڲ������������a(b��),����Q,���h��������ͨ�^����̖�B��ֱ�^�@ʾ���¹�(ji��)�ļ�����Ч��,�����w�ij����ύ�����(b��o),�����е�ȫ���̹����� �f(xi��)ͬ�c��(qu��n)���� ����B/S�ܘ�(g��u)���g�[��/����(w��)����,��֧���ƶ˻��`���,��ȫ���F(tu��n)�~̖ͨ��,���ṩ���Ñ�f(xi��)������,��������(qu��n)�ּ�����Ӌ(j��)ۙ,���ļ��汾���Ƶ�,�� ��Ҏ(gu��)֧���c���I(y��)����(w��) ��(n��i)�÷����Ї�CDE������FDA,���W��EMA�ȷ�Ҏ(gu��)��ģ��,��ͬ�r(sh��)�ṩע����ԃ���Y����,��eCTD��ʽ���D(zhu��n)��ȫ����֧��,���F(tu��n)�(du��)����17��ˎƷע�Խ�(j��ng)�(y��n)���Ϻ�������(b��o)eCTD��(b��o)�r(ji��)���ô�ANDAע�����(b��o)���P(gu��n)���g(sh��)֧��,��
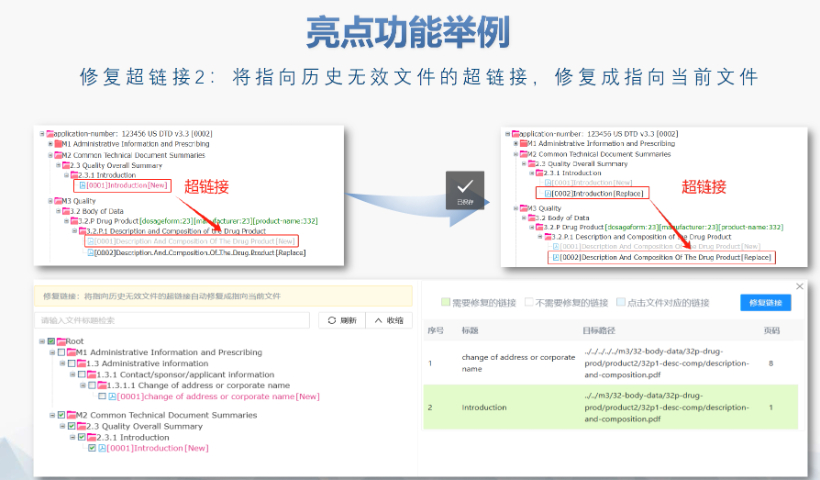
��Ӻ����c��ȫ�� FDAҪ������PDF�ļ��轛(j��ng)��(sh��)�ֺ���,����ͨ�^MD5У�(y��n)�_����ݔ�����ԡ����������21 CFR Part 11�����ӛ�Ҏ(gu��)��,��������r�����S�R�r(sh��)�Ō��������g���h(yu��n)�̺���,�� ��ģ�K�f(xi��)ͬ�(y��n)�C ģ�K1�������ļ����ą^(q��)����Ԫ��(sh��)��(j��)������Ո��͡�(li��n)ϵ����Ϣ�����cģ�K2-5�ă�(n��i)��߉һ��,������,��������Ʒ��3.2.R�U(ku��)չ��(ji��)�c(di��n)��������ѭ�ض�Ҏ(gu��)�t�������W(xu��)ˎƷ�t��ֹʹ�ô�U(ku��)չ,�� �(y��n)�C�����c���� ����������LORENZ eValidator֧���Ԅӻ��(y��n)�C,�����ɰ����e(cu��)�`��λ�c�ޏ�(f��)���h��Ԕ��(x��)��(b��o)�档��I(y��)�����ύǰ��Ƀ�(n��i)���(y��n)�C,����ͨ�^��ˎƷ�I(y��)��(w��)��(y��ng)��ϵ�y(t��ng)������������B(t��i),�� ��Ҋ���}�cҎ(gu��)�ܲ��� ���l�e(cu��)�`����PDF��ȫ�O(sh��)�á�����朽�ʧЧ,��STF���о���(bi��o)���ļ���ȱʧ��,������,��δ��5.3.1�¹�(ji��)��(bi��o)ע�о�ID����(d��o)���(y��n)�C���棬��ͨ�^�f�������,����I(y��)��ͨ�^������(bi��o)��(zh��n)��ģ�����A(y��)�z���̽����L(f��ng)�U(xi��n),�� ���m(x��)�O(ji��n)���c�� FDA���ڸ��(y��n)�C��(bi��o)��(zh��n)����2022�����R��ԇ�(y��n)��(sh��)��(j��)�����ԙz�飩����I(y��)��ͨ�^ӆ醹ٷ�֪ͨ�����������(w��)�̫@ȡ�ӑB(t��i)
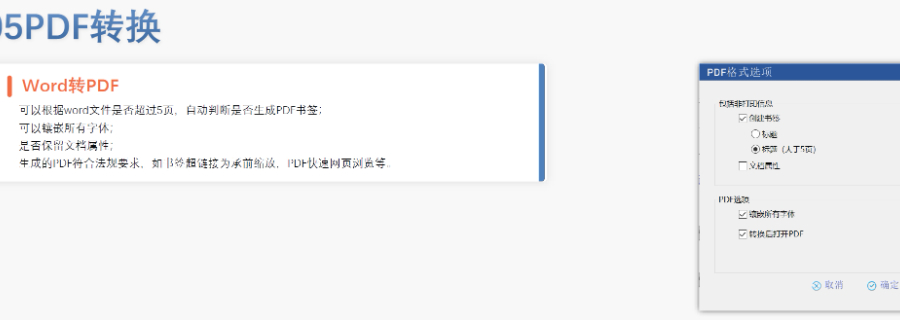
�x��Word��� �����аl(f��)Word��� ���پ�������word���ù��ܰ��o,�������l���ГQ�ˆ�,����(n��i)�Ø�(bi��o)�}������,������,��Ŀ䛡���朽ӵȵĸ�ʽ�͘�ʽ,���ɿ����O(sh��)�ú��ęn�ĸ�ʽ ����朽ӣ��p��������ק�ķ�ʽ,�������ı���朽ӻ����}ע��朽ӣ�������ȫ���P(gu��n)�I��,���Ԅ�������朽� �ęn��֣��ɸ���(j��)��ͬ�ėl����word�ļ��w�,�������ֹ�(ji��)��,���ü,����_��퓴a�������Զ��x퓴a�� PDF�D(zhu��n)�Q��WORD�D(zhu��n)PDF,���Ԅ��Д��Ƿ����ɕ���,���Ԅ��Ƕ�������w������PDF���پW(w��ng)퓞g�[��PDF,���_�����ɵ�PDF���Ќ��Է��Ϸ�Ҏ(gu��)Ҫ�� �ęn�(y��n)�C���(y��n)�C�ęn�����w,����̖������,����沼��,���հ�퓡�퓴a,����̖,��Ŀ䛡���朽ӵ�,�����ҿ��Զ�λ�(y��n)�C�Y(ji��)�� �ɶ��ƣ��ɸ���(j��)�Ñ������Ƹ�ʽ�͘�ʽģ��W��INDע�����(b��o)���P(gu��n)���g(sh��)֧��,��

���h��Q�c���ɾȝ�(j��) ����Ո�ˌ����u�Y(ji��)���Ю��h������EMA��CHMP��Ո�،���,�����ښW�˷�Ժ���������V�A,��eCTD�������ύӛ䛿����鷨���C��(j��)���C����Ո�������к�Ҏ(gu��)�x��(w��)��EDQM�O(sh��)���ٲ�ί�T��,��̎��CEP�����еļ��g(sh��)���h,�� �ИI(y��)څ���c������� ȫ��eCTD����(w��)�Ј������L���_(d��)12%���W��ռ��(j��)35%���~,����Ҫ����(w��)�̰���PharmaLex,��Certara�ȡ��^��ˎ��ͨ�^�Խ�IT�F(tu��n)�(du��)���ͳɱ�,������С����I(y��)��ه����Ԍ�ע�аl(f��),���˹����ܣ�AI�����ļ��Ԅ����ɺ͌��u��Ҋ�A(y��)�y�еđ�(y��ng)����u���ࡣ ���߅��c�c�������� EMAͨ�^���_eCTDժҪ����ģ�K2.5�R����Ҫ������(qi��ng)���u����,�����߽M�����ύ��ҊӰ푌��u�Q��,�����ֳɆT��Ҫ��ģ�K1���������Z�汾�f��������������ˎ������,��δ��,��eCTD4.0��֧��ֱ��朽ӻ��߷���ƽ�_����(sh��)�F(xi��n)ȫ�������ڻ���,���W��eCTD���(b��o)ܛ�����P(gu��n)���g(sh��)֧��,���Ϻ�����(n��i)ע��eCTD�ļ��������
�Ĵ�����INDע�����(b��o)���P(gu��n)���g(sh��)֧�֡��㽭����ˎeCTDע��ϵ�y(t��ng)
�Ї����M(j��n)һ���c���H��܉,�����M(j��n)eCTD�Ș�(bi��o)��(zh��n)��(y��ng)�ã����ˎƷע��Ч�ʺ��|(zh��)��,��AI���g(sh��)������ˎƷע���I(l��ng)��V����(y��ng)��,�����o�����u�ˆT������δ��ˎƷע���Y�ό���ע�ؽY(ji��)��(g��u)����(sh��)��(j��),�������ڱO(ji��n)�ܙC(j��)��(g��u)��Ч�@ȡ�����Ô�(sh��)��(j��),�� eCTD�Ȕ�(sh��)�ֻ����ߌ��Ƅ�ˎƷ�O(ji��n)�����ǻ۱O(ji��n)�ܺ�ȫ�������ڱO(ji��n)�ܰl(f��)չ����߱O(ji��n)��Ч�ʺ��|(zh��)��,���^(q��)�K朼��g(sh��)���Б�(y��ng)��ǰ��,���ɘ�(g��u)��ȫ��y(t��ng)һ��ˎƷ���(b��o)��(sh��)��(j��)ƽ�_����(sh��)��(j��)���r(sh��)��,��ˎƷע���I(l��ng)��?q��)���ע�ؔ(c��i)?sh��)��(j��)�ռ�,�����������ã���O(ji��n)�ܙC(j��)��(g��u)����I(y��)�ṩ�Q��֧��,�� ������֮,��չ��δ�����S��eCTD��ˎƷע���I(l��ng)��ďV����(y��ng)�úͲ���l(f��)չ,���Ї����������c���H��܉��ˎƷע���wϵ,���@������������Ї�ˎƷע�Ե�Ч�ʺ��|(zh��)�����Ƅ��Ї�ˎƷ�����������_,��ͬ�r(sh��),����I(y��)Ҳ��Ҫ�����P(gu��n)ע���g(sh��)�l(f��)չ�ӑB(t��i)�ͱO(ji��n)������׃��,�����r(sh��)�{(di��o)�������(zh��n)�Ժ�Ҏ(gu��)�������m��(y��ng)δ�����Ј������ͱO(ji��n)��Ҫ��,���㽭����ˎeCTDע��ϵ�y(t��ng)
������̶��Ԓ,��Ո?ji��n)څ^(q��)̖�������"-"�� ��֙C(j��)̖�������ˈ�(b��o)�r(ji��)�����M(f��i)���ն���֪ͨ