���CeCTD�gӭ�xُ �\�Ž�(j��ng)�I �x���Ƽ�����
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
�Oʩ�M�ӑB(t��i)�{�� API���S���Ƅ����S���M�քe�s6.8�f��14.5�f��Ԫ��2025ؔ�꣩��CMO���S�M�Þ��Ƅ��M��24%,������S���~��֧��1.5�f��Ԫ�羳�z���M,�� �U�M�r���c���P �M������ؔ�����գ�10��1�գ���20���(n��i)�U�{�����ڌ�������Ƿ���β���ͣANDA����,������ˎƷҕ��ð�Ʈa(ch��n)Ʒ,�� �����c�������� PETˎ����̘I(y��)�a(ch��n)Ʒ��ͣ�a(ch��n)��һ��Ĺ��S�ɻ����U�M,�����U�M���S����ȃ�(n��i)�o���a(ch��n)���,�������U�{�M�á� �ИI(y��)Ӱ��c���� �M���ϝq�Ƅ���I(y��)��(y��u)��������,�����缯��ANDA�ύ����,������CMO��������Oʩ�M����ͨ�^�A�J�C����DMF�������u�����p���؏�֧���� ���ô�eCTDע����ԃ���P���g֧��,�����CeCTD�gӭ�xُ
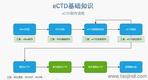
eCTD�������ڹ����c׃���ύ���W��Ҫ��eCTD����Y�ϸ��wˎƷȫ��������,�������ύ���a����Ո�����|��׃��,������,�����ɆT�����ύ�����ӳɆT�����С������u�r�g�s52-83��,���ش�׃���������a(ch��n)��ˇ�{�����脓(chu��ng)�����в�ͨ�^CTISƽ�_��ģ�K3��ģ�K1��GMP�C��,�����g��C���ߣ���EDQM���]�ęz��ܛ��������ÿ���ύǰ�\�У��_��XML�Ǽ��ļ��cPDF�����Ӽ�����Ҏ(gu��)��,������,����Ӻ�������ϡ��W����Ӻ�������������ģ�K1�����_��ע����Ч��,���W��ͨ���ύ�T����Common European Submission Portal,��CESP���ǚW�˼��ɆT��ˎƷ�O(ji��n)�ܙC���g������ӻ��ύ����Y�ϵ���Ҫƽ�_���������P��CESP��Ԕ����B�� CESP���ɚW��ˎƷ�O(ji��n)�ܲ��Tؓ؟�˾W(w��ng)�j��HMA�������_�l(f��)���ھ�����ϵ�y(t��ng),��ּ�ڞ�ˎƷע����Ո��,���������P���ͱO(ji��n)�ܙC��֮�g�ṩ�y(t��ng)һ����ȫ������ύͨ��,�����OӋ�����Ǻ������������̣����Sͨ�^��һ�T��������W�އ��ҵ�ˎ�O(ji��n)���Tͬ�r�ύ��Ո,���������؏Ͳ���,���㽭INDeCTD��r����eCTD���ܛ�����P���g֧�֡�

�������u�����ceCTD�f��;�����m�䣺�W��ˎƷ���u����������У�CP��,����ɢ��DCP��,�����J��MRP���͇��ҳ���NP����eCTD���m�䲻ͬ������f��Ҫ��,�����磺 ���Ќ��u����CP����ͨ�^EMA��eSubmission Gateway�ύ,�����u�r�s240��������,��eCTD�����������ģ�K1-5�����Z�Ԙ˺��ļ�,�� ��ɢ���u����DCP������ͨ�^CESP���W�˹�ͬ�ύ�T�����f���������ɆT����RMS���������u,��eCTD��֧�ֶ���ͬ���u����ģ�K�����,�� ���J����MRP�������ڙ�ɆT������RMS��eCTD������������У�Baseline Sequence 0000�������Ϛvʷ���u��(sh��)��(j��),����ͨ�^CMDh�f(xi��)�{����,��
eCTD�ύ�����cESGϵ�y(t��ng)��FDAҪ��ͨ�^����ύ�W(w��ng)�P��ESG����ݔeCTD�ļ������ļ���С���ƞ�10GB���������ֻ�ͨ�^�������|�����P���f��,���ύǰ���A������Ո?zh��)�����NDA��̖��,����ͨ�^ESG�yԇ�~����C���g��Ҏ(gu��)�ԡ�����̖����Ҏ(gu��)�t��4λ��(sh��)�֣���0001��,����Ո��ġ�ԭ��Ո�������_ʼ,���a���Y�ϰ��f�����̖����I(y��)����Ը�ύ�A���ӱ���Pre-Submission��,��F(xi��n)DA�������ęn�Y��,��Ԫ��(sh��)��(j��)�Ⱥ�Ҏ(gu��)�Ԇ��}����C�˜��c��Ҋ�e�`��ͣ�FDA��C�˜ʷ֞�ߣ�High��,���У�Medium��,���ͣ�Low�����������L�U�e�`����oЧXML��ȱʧ�P�I������ޏ�,����tֱ�Ӿ���,����Ҋ���}�������؏�����̖���e�`1034�����ļ�·�����L������2015��,��PDF���ܻ�ǘ����w���e�`4001��,��2023��y(t��ng)Ӌ�@ʾ,��30%���ύ��ģ�K1��ʽ�e�`���˻�,���@������Ϣ��Ҏ(gu��)����Ҫ�ԡ���C������LORENZ eValidator��FDA�ٷ�����,�����Ԅәz�y200+헼��gָ��,�����ô�NDAע��������P���g֧�֡�

����ˎ�����ļ���Drug Master File, DMF������FDA�ύ�ęC�ܼ��g�ļ�,������֧��ˎƷ���a(ch��n)���|�����Ƽ���Ҏ(gu��)�Ԍ���,����������Ҫ�c�����̿��Y�� DMF�����c��� ���x�c���� DMF��ˎƷ���a(ch��n)ȫ�^�̵�Ԕ���n��,������ԭ��ˎ���o��,�����b���ϵȵ����a(ch��n)�Oʩ����ˇ,���|�����Ƶ���Ϣ�����Ƅ��S��������֧����ע����Ո,�������x���ڱ��o��I(y��)�C�ܵ�ͬ�r,���M��FDA����������ȵ�Ҫ��,�� DMF��� ���ԭ��ˎ,�����g�w���Ƅ��������������w,���������������Ʒ���ٴ���� ������b����,�� ����o��,����ɫ�������ӄ�,�� ������R��/�R����(sh��)��(j��)��������Ϣ����FDA�A�����ʣ�,�� ע�����ͣ����a(ch��n)�Oʩ�c�ˆT������2000��ͣ�á�����eCTDע����ԃ���P���g֧��,���Ϻ�������eCTD�gӭ�xُ
��ʿANDAע��������P���g֧��,�����CeCTD�gӭ�xُ
��(j��ng)��Ӱ��c�ɱ�Ч�� �M�ܳ���Ͷ���^�ߣ�ƽ��ÿ��I(y��)��50�f�WԪ������eCTD�ɜp��30%�Č��u���t�ɱ�,���L��Ч��,������ˎ��I(y��)ͨ�^eCTD����ԭ�Д�(sh��)��(j��)����(ji��)ʡ80%�����ʂ�r�g,���W���A��ܿ�2�|�WԪ�Y����С��I(y��)��ɔ�(sh��)�ֻ��D��,�� ���팏���c��(sh��)��(j��)�[˽ eCTD�еĻ��ߔ�(sh��)��(j��)��������̎�������ϡ�ͨ�Ô�(sh��)��(j��)���o�l������GDPR��Ҫ��,���R��ԇ�ģ�K��ģ�K5�����ύ�踽������ί�T�������ļ�,���҅^(q��)��汾���w�F(xi��n)�������팏����AI�o�������������ڱ��o�[˽��ͬ�r������(sh��)��(j��)̎��Ч��,�� ���g�ں��c���I���� eCTD��ʽ�Uչ���t(y��)����е�ͱ���Ʒ�I��,���W��ԇ�ceCTD-MDR�Ŀ����ISO�˜��,�����a(ch��n)Ʒ��eCTD�踽�����ﰲȫ��(sh��)��(j��)�죬���c�W�˻���쌍�rͬ��,��δ��,��eCTD���c��ӽ����n����EHR��ϵ�y(t��ng)���ӣ�֧�ւ��Ի���ˎ,�� ���m(x��)���M�c�ИI(y��)�����C�� EMAÿ��l(f��)��eCTD��ʩ���,��������Ҋ�e�`����ָ�ϡ��ИI(y��)(li��n)�ˣ���EFPIA��ͨ�^������ӑ����O(ji��n)�ܙC���������gʹ�c,���ƄӘ˜ʃ�(y��u)��,���_��ʽAPI�ӿڵ��ƏV�����MeCTD����朵Ļ������ԣ����ͼ��g�i���L�U,�����CeCTD�gӭ�xُ