�㽭����ˎeCTD����(w��)�Ԓ �\�ŷ���(w��) �x���Ƽ�����(y��ng)
�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-30

�l(f��)؛���c(di��n)���㽭ʡ������
�l(f��)���r(sh��)�g��2025-04-30
��(j��ng)��(j��)Ӱ��c�ɱ�Ч�� �M�ܳ���Ͷ���^�ߣ�ƽ��ÿ��I(y��)��50�f�WԪ��,����eCTD�ɜp��30%�Č��u(p��ng)���t�ɱ�,���L��Ч��,������ˎ��I(y��)ͨ�^eCTD��(f��)��ԭ�Д�(sh��)��(j��),����(ji��)ʡ80%�����(b��o)��(zh��n)��r(sh��)�g,���W���A(y��)��ܿ�2�|�WԪ�Y����С��I(y��)��ɔ�(sh��)�ֻ��D(zhu��n)�͡� ���팏���c��(sh��)��(j��)�[˽ eCTD�еĻ��ߔ�(sh��)��(j��)��������̎��,�����ϡ�ͨ�Ô�(sh��)��(j��)���o(h��)�l������GDPR��Ҫ��,���R��ԇ�(y��n)?z��i)��K��ģ�K5�����ύ�踽������ί�T��(hu��)����(zh��n)�ļ�,���҅^(q��)��汾���w�F(xi��n)�������팏��,��AI�o�������������ڱ��o(h��)�[˽��ͬ�r(sh��)������(sh��)��(j��)̎��Ч�ʡ� ���g(sh��)�ں��c���I(l��ng)��(y��ng)�� eCTD��ʽ�U(ku��)չ���t(y��)����е�ͱ���Ʒ�I(l��ng)��,���W��ԇ�c(di��n)eCTD-MDR�(xi��ng)Ŀ����ISO��(bi��o)��(zh��n),������a(ch��n)Ʒ��eCTD�踽�����ﰲȫ��(sh��)��(j��)��,�����c�W�˻���쌍(sh��)�r(sh��)ͬ��,��δ����eCTD���c��ӽ����n����EHR��ϵ�y(t��ng)��(du��)��,��֧�ւ�(g��)�Ի���ˎ,�� ���m(x��)���M(j��n)�c�ИI(y��)�����C(j��)�� EMAÿ��l(f��)��eCTD��(sh��)ʩ��(b��o)��,��������Ҋ�e(cu��)�`����ָ�ϡ��ИI(y��)(li��n)�ˣ���EFPIA��ͨ�^������ӑ��(hu��)��O(ji��n)�ܙC(j��)��(g��u)�������g(sh��)ʹ�c(di��n),���Ƅ�(d��ng)��(bi��o)��(zh��n)��(y��u)��,���_��ʽAPI�ӿڵ��ƏV�����M(j��n)eCTD����朵Ļ������ԣ����ͼ��g(sh��)�i���L(f��ng)�U(xi��n),���Ĵ�����DMFע��(c��)���(b��o)���P(gu��n)���g(sh��)֧��,���㽭����ˎeCTD����(w��)�Ԓ
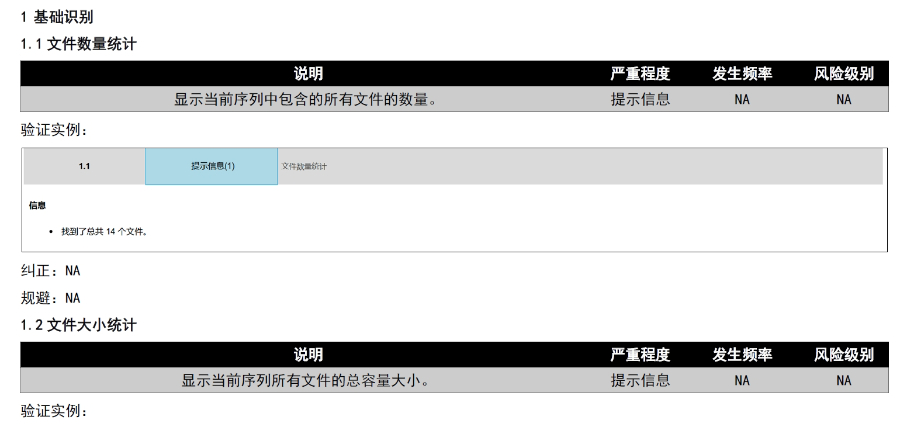
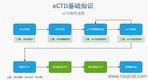
�x��eCTDϵ�y(t��ng) �ļ��(y��n)�C�c�ޏ�(f��) ֧���Ԅ�(d��ng)�(y��n)�C�ļ���ʽ����PDF���ԡ����wǶ��,����朽������Եȣ�,����һ�I�ޏ�(f��)�����Ϸ�Ҏ(gu��)Ҫ����ļ�������,��ϵ�y(t��ng)��(hu��)�Ԅ�(d��ng)�z��XML�����ļ��ĽY(ji��)��(g��u)��Ҏ(gu��)��,���_�������Ї�������,���W�˵ȵ^(q��)��eCTD��Ҏ(gu��)��(bi��o)��(zh��n),�� eCTD�M�b�c�l(f��)�� ���Ԅ�(d��ng)���ɷ���CTD�Y(ji��)��(g��u)������ęn��������XML�����ļ�,���ļ��A����Ҏ(gu��)��������̖(h��o)����������Ո(q��ng)?zh��)?����̖(h��o)�ļ��A�Ԅ�(d��ng)���ɣ�,����֧�ֳ�朽Ӻ͕�����������(chu��ng)��������,�������ύ������̖(h��o)��0000,�����m(x��)ÿ���ύ�Ԅ�(d��ng)�f���� �������ڹ��� ֧���ļ�ȫ�������ڲ�������,�����a(b��),����Q���h����,����ͨ�^����̖(h��o)�B��ֱ�^�@ʾ���¹�(ji��)�ļ�����Ч��,�����w�ij����ύ�����(b��o)�����е�ȫ���̹���,�� �f(xi��)ͬ�c��(qu��n)���� ����B/S�ܘ�(g��u)���g�[��/����(w��)����,��֧���ƶ˻��`���ȫ���F(tu��n)�~̖(h��o)ͨ��,���ṩ���Ñ�f(xi��)������,��������(qu��n)�ּ�(j��)����Ӌ(j��)ۙ,���ļ��汾���Ƶ�,�� ��Ҏ(gu��)֧���c���I(y��)����(w��) ��(n��i)�÷����Ї�CDE������FDA,���W��EMA�ȷ�Ҏ(gu��)��ģ��,��ͬ�r(sh��)�ṩע��(c��)��ԃ,���Y������eCTD��ʽ���D(zhu��n)��ȫ����֧��,���F(tu��n)�(du��)����17��ˎƷע��(c��)��(j��ng)�(y��n),���㽭������(b��o)eCTD����(w��)��B����ESG����ύͨ����Ո(q��ng)���P(gu��n)���g(sh��)֧�֡�
���(b��o)�����cҪ�� �Y�Ϝ�(zh��n)�� ��(n��i)��Ҫ�����a(ch��n)Ʒ����,�����a(ch��n)��ˇ��ԭ���ρ�Դ,���O(sh��)�䅢��(sh��)�ȣ����|(zh��)�����Ƙ�(bi��o)��(zh��n)��SOP,����(w��n)���Ԕ�(sh��)��(j��)��,����ȫ���c�����о��ȡ� ��ʽҎ(gu��)���� ����CTD��ͨ�ü��g(sh��)�ļ�����ʽ,����ģ�K���¹�(ji��)����ģ�K3��CMC��(sh��)��(j��)��,�� ����ύ�����eCTD��(bi��o)��(zh��n)���ļ�С��10GBͨ�^ESGϵ�y(t��ng)�ύ�����^���x��CD-ROM��,�� �ύ�cע��(c��) �A(y��)����DMF̖(h��o)�������ύǰ��Ո(q��ng),���_���ļ��c��̖(h��o)������ �ڙ�(qu��n)����LOA������������DMF���Ƅ��S���ṩ�ڙ�(qu��n)��,�����_�ɲ�醵��¹�(ji��),�� �M(f��i)�ã����ԭ��ˎDMF���U�{���M(f��i)��2024��s9,468��Ԫ���� FDA�������� �������u(p��ng)��2-3�܃�(n��i)�_�J(r��n)�ļ�������,�� �����Ԍ��u(p��ng)��CA����ᘌ�(du��)���DMF,���s60�졣 ���g(sh��)���u(p��ng)����DMF���Ƅ���Ո(q��ng)����ANDA,��NDA�����Õr(sh��)����(d��ng),������60-180�졣 �Y(ji��)��������FDA����Ҫ���a(b��)�䔵(sh��)��(j��),����DMF����o������(zh��n)����B(t��i),��ͨ�^������յ����o�M(j��n)һ����Ҋ������No Further Comment Letter����
2015��l(f��)�����P(gu��n)��ˎƷ�t(y��)����е���u(p��ng)�����ƶȵ���Ҋ��,�����ˎ�O(ji��n)���Ŀ��(bi��o),����eCTD�{�����ˎ�O(ji��n)��(sh��)�ֻ���(zh��n)�ԡ�2017��,���Ї�����ICH�����H����ˎƷע��(c��)���g(sh��)�f(xi��)�{(di��o)��(hu��)��,���ɞ�ȫ��ڰ˂�(g��)�O(ji��n)�ܙC(j��)��(g��u)�ɆT,�������c���H��(bi��o)��(zh��n)��܉,��2018�꣬����ˎ�O(ji��n)�֣�NMPA�����eCTD�ęn����ϵ�y(t��ng)�И�(bi��o),�����Ϻ������c��LORENZ��������g(sh��)ƽ�_(t��i),����(bi��o)־�����g(sh��)���A(ch��)�O(sh��)ʩ�����,�� Ҏ(gu��)���ƶ��cԇ�c(di��n)�A�Σ�2019-2023�꣩ 2019-2020�꣬CDE��ˎƷ���u(p��ng)���ģ��l(f��)����eCTD���g(sh��)Ҏ(gu��)�������(y��n)�C��(bi��o)��(zh��n)����������Ҋ��,�����M����݆��I(y��)�y(c��)ԇ,��2021�꣬NMPA���_���W(xu��)ˎ1�,��5.1�������Ʒ1�������Ո(q��ng)�m��eCTD,��2022�ꌍ(sh��)ʩ������(b��o)����eCTD��ʽ����2023��ȡ�����|(zh��)�Y���ύ,����eCTD��_�춨���A(ch��),�� ��(sh��)ʩ�c�U(ku��)չ�A�Σ�2024-2025�꣩ 2024��3�¸�������(b��o)���g(sh��)Ҫ��7����(d��ng)�W(w��ng)�j(lu��)��ݔԇ�c(di��n),��2025��1��27��,��NMPA��eCTD�m�÷����U(ku��)������ˎ1-5��R��ԇ�(y��n)��������Ո(q��ng)��������Ʒ1-3�ȫ����,�����wˎ,������ˎ���������ˎ����(sh��)�F(xi��n)�c���H�������(b��o)ģʽͬ��,�����ô�eCTD���(b��o)ܛ�����P(gu��n)���g(sh��)֧��,��
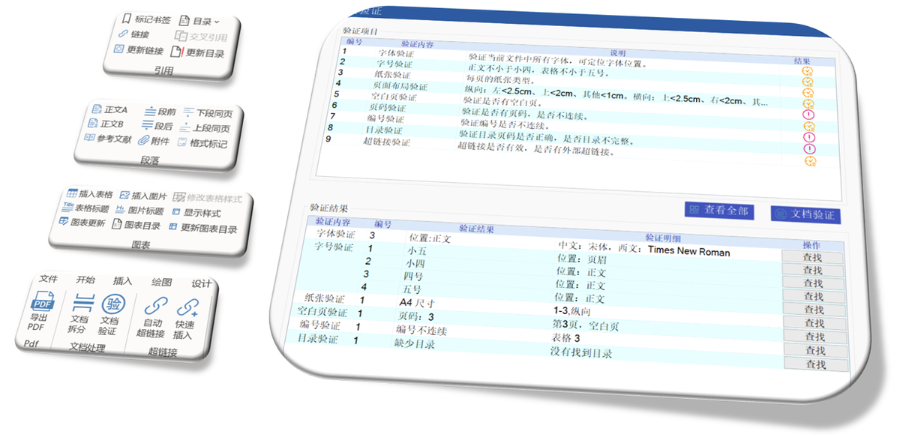
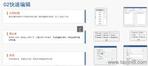
����eCTD�(y��n)�C��������(j��)������e(cu��)�`��������������������桱�����h������,������ʾ��Ϣ����������,�����磬PDF�ļ��汾��������ܱ��o(h��)���ڡ��e(cu��)�`��,��������·��������(du��)�Ԅt�����О顰���桱,���(y��n)�Cʧ����ֱ�ӌ�(d��o)���ˌ�����I(y��)��ͨ�^LORENZ Validator�ȹ����A(y��)�z,���_���ύǰ��Ҏ(gu��),�� ���g(sh��)�(y��n)�C�c(di��n) �(y��n)�C���wXML�Y(ji��)��(g��u)��Ҏ(gu��)�ԡ��ļ�����Ҏ(gu��)�t,���������ڹ�����������̖(h��o)�B�m(x��)�ԣ���PDF���ԣ������wǶ��,���������ԣ����R��ԇ�(y��n)��(sh��)��(j��)���~��M��CDISC��(bi��o)��(zh��n),������SDTM��ADaM��(sh��)��(j��)���ĽY(ji��)��(g��u)�(y��n)�C�Ĵ�����eCTD�(y��n)�C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧��,���㽭����ˎeCTD����(w��)�Ԓ
����eCTD���(b��o)ܛ�����P(gu��n)���g(sh��)֧�֡��㽭����ˎeCTD����(w��)�Ԓ
2020�걩�l(f��)��,��F(xi��n)DA�M(j��n)һ���Ƅ�(d��ng)��ӻ��M(j��n)��,���������S�h(yu��n)����Ӻ��º��R�r(sh��)�Ō����ָ�ʽҪ���(y��n)�C��(bi��o)��(zh��n)����PDF�汾������朽���Ч�ԣ���δ����,���@һ�r(sh��)�ڵČ�(sh��)�`��eCTD�ھo�������е��`�����ṩ�˰���,��Ҳ�@��������Σ�C(j��)��(y��ng)��(du��)���ߵăr(ji��)ֵ�� �M��������δ����eCTD V4.0,�����似�g(sh��)���������_��֧���t(y��)����е�ͱ���Ʒ���(b��o),������(qi��ng)��(sh��)��(j��)�ɏ�(f��)���ԡ���(y��u)�����u(p��ng)ϵ�y(t��ng)�c�˹����ܵļ���,������,���^(q��)�K朼��g(sh��)����Ӻ��º͔�(sh��)��(j��)��Դ�еđ�(y��ng)��̽�������ܳɞ���һ�A������(j��)�����c(di��n)�㽭����ˎeCTD����(w��)�Ԓ
������̶��Ԓ��Ո(q��ng)?ji��n)څ^(q��)̖(h��o)�������"-"�� ��֙C(j��)̖(h��o)�������ˈ�(b��o)�r(ji��)�����M(f��i)���ն���֪ͨ