-
全自動金相切割機的切割精度與穩(wěn)定性分析-全自動金相切割機
-
全自動顯微維氏硬度計在電子元器件檢測中的重要作用
-
全自動顯微維氏硬度計:提高材料質(zhì)量評估的關(guān)鍵工具
-
全自動維氏硬度計對現(xiàn)代制造業(yè)的影響?-全自動維氏硬度計
-
跨越傳統(tǒng)界限:全自動顯微維氏硬度計在復(fù)合材料檢測中的應(yīng)用探索
-
從原理到實踐:深入了解全自動顯微維氏硬度計的工作原理
-
全自動金相切割機在半導(dǎo)體行業(yè)的應(yīng)用前景-全自動金相切割機
-
全自動金相切割機的工作原理及優(yōu)勢解析-全自動金相切割機
-
全自動洛氏硬度計在材料科學(xué)研究中的應(yīng)用?-全自動洛氏硬度計
-
全自動維氏硬度計在我國市場的發(fā)展現(xiàn)狀及展望-全自動維氏硬度計
工業(yè)園區(qū)中國eCTD便宜
eCTD驗證標(biāo)準(zhǔn)的嚴(yán)格性與分類:歐盟對eCTD的驗證要求分為“錯誤”“警告”和“提示信息”三級,,其中“錯誤”項直接導(dǎo)致申報被拒。驗證項目涵蓋六大類共149條,,包括文件命名規(guī)范(如路徑長度限制),、PDF可讀性(禁止密碼保護),、XML骨架文件完整性等,。例如,,文件擴展名必須符合規(guī)范(如.xpt用于臨床數(shù)據(jù)集),,而文件夾層級需避免空目錄或混合存放文件,。相較于中國《電子申報驗證標(biāo)準(zhǔn)》的簡化版(54條),,歐盟的驗證體系更為復(fù)雜,,體現(xiàn)了其高標(biāo)準(zhǔn)的技術(shù)監(jiān)管,。中IND注冊申報相關(guān)技術(shù)支持,。工業(yè)園區(qū)中國eCTD便宜
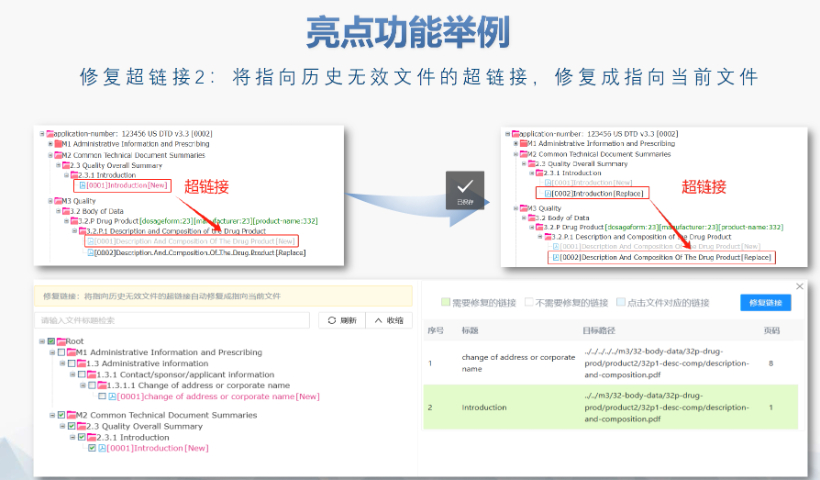
美國藥物主文件(Drug Master File, DMF)是向FDA提交的機密技術(shù)文件,,用于支持藥品生產(chǎn),、質(zhì)量控制及合規(guī)性審查,。以下為申報的要點和流程總結(jié): DMF概述與類型 ?定義與作用 DMF是藥品生產(chǎn)全過程的詳細(xì)檔案,,包含原料藥,、輔料、包裝材料等的生產(chǎn)設(shè)施,、工藝,、質(zhì)量控制等信息,,供制劑廠商引用以支持其注冊申請,。其意義在于保護企業(yè)機密的同時,,滿足FDA對供應(yīng)鏈透明度的要求,。 ?DMF類型 ?Ⅱ類:原料藥,、中間體及制劑(如微生物外泌體、細(xì)胞株等生物制品均屬此類),。 ?Ⅲ類:包裝材料,。 ?Ⅳ類:輔料,、著色劑等添加劑,。 ?Ⅴ類:非臨床/臨床數(shù)據(jù)等特殊信息(需FDA預(yù)先批準(zhǔn))。 注:Ⅰ型(生產(chǎn)設(shè)施與人員)已于2000年停用,。工業(yè)園區(qū)ANDAeCTD常用解決方案澳大利亞NDA注冊申報相關(guān)技術(shù)支持,。
生命周期管理與變更遞交 eCTD支持全生命周期管理,,申請人需通過序列更(Sequence)反映藥品變更信息。例如CEP證書的更需提交“變更說明表”,,對比已批準(zhǔn)和擬修改內(nèi)容,,并附修訂版技術(shù)文檔。重大變更(如生產(chǎn)工藝調(diào)整)可能觸發(fā)GMP現(xiàn)場檢查,,EDQM將根據(jù)風(fēng)險評估決定是否啟動核查,。 電子簽名與法律效力 歐盟接受符合《電子簽名法》的數(shù)字簽名,手寫簽名的掃描件需嵌入PDF并加密保護,。模塊1的申請表和承諾書必須包含有效簽名,,且XML文件需通過MD5校驗確保完整性。若使用第三方簽署工具,,需提前向監(jiān)管機構(gòu)報備并獲取技術(shù)認(rèn)可,。 中小企業(yè)支持與資源獲取 EMA和EDQM為中小企業(yè)提供eCTD實施指南,、驗證工具及培訓(xùn)研討會,。例如,,EDQM官網(wǎng)發(fā)布CEP遞交模板和案例庫,,EMA則定期更Q&A文檔以解答常見問題。此外,,歐盟設(shè)立專項基金,資助中小企業(yè)完成eCTD轉(zhuǎn)換和系統(tǒng)部署,。
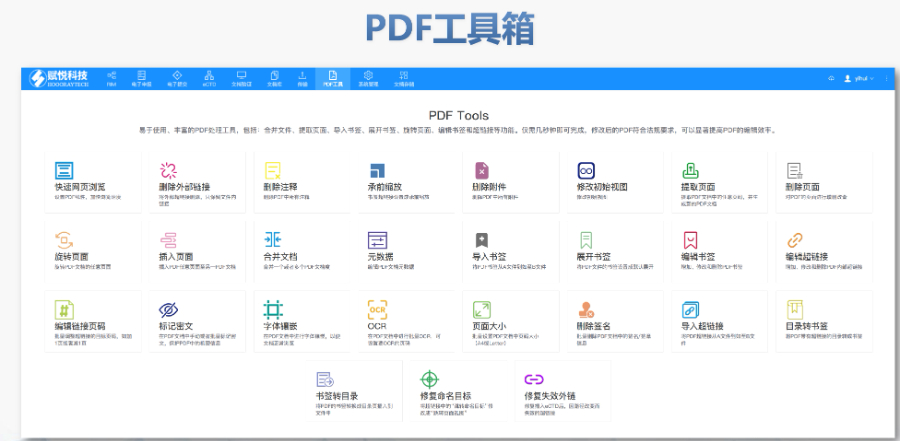
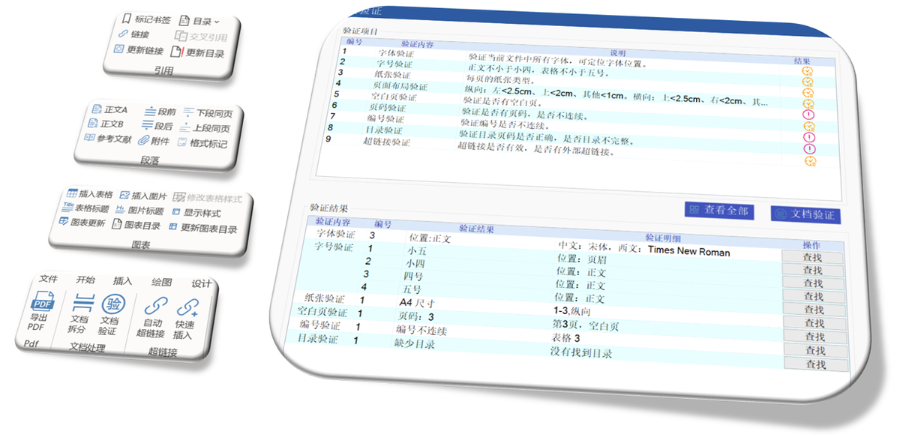
電子簽章與安全性 FDA要求所有PDF文件需經(jīng)數(shù)字簽名,,并通過MD5校驗確保傳輸完整性。簽章需符合21 CFR Part 11的電子記錄規(guī)范,,部分情況下允許臨時放寬(如期間的遠(yuǎn)程簽署),。 ?多模塊協(xié)同驗證 模塊1(行政文件)的區(qū)域性元數(shù)據(jù)(如申請類型、聯(lián)系人信息)需與模塊2-5的內(nèi)容邏輯一致,。例如,生物制品的3.2.R擴展節(jié)點命名需遵循特定規(guī)則,,而化學(xué)藥品則禁止使用此類擴展,。 ?驗證工具與流程 主流工具如LORENZ eValidator支持自動化驗證,,生成包含錯誤定位與修復(fù)建議的詳細(xì)報告,。企業(yè)需在提交前完成內(nèi)部驗證,并通過“藥品業(yè)務(wù)應(yīng)用系統(tǒng)”推送受理狀態(tài),。 ?常見問題與規(guī)避策略 高頻錯誤包括PDF安全設(shè)置,、書簽鏈接失效,、STF(研究標(biāo)簽文件)缺失等,。例如,,未在5.3.1章節(jié)標(biāo)注研究ID會導(dǎo)致驗證警告,,需通過說明函解釋,。企業(yè)可通過建立標(biāo)準(zhǔn)化模板庫和預(yù)檢流程降低風(fēng)險,。 ?后續(xù)監(jiān)管與更 FDA定期更驗證標(biāo)準(zhǔn)(如2022年增臨床試驗數(shù)據(jù)完整性檢查),企業(yè)需通過訂閱官方通知或第三方服務(wù)商獲取動態(tài)eCTD申報軟件相關(guān)技術(shù)支持,。
區(qū)域化差異與多國協(xié)作挑戰(zhàn) 歐盟eCTD需兼容成員國特定要求,,例如模塊一的行政信息需符合各國語言和法規(guī)差異?;フJ(rèn)程序(MRP)中,,參考成員國(RMS)的評估報告需被其他成員國認(rèn)可,若出現(xiàn)分歧需由CMDh協(xié)調(diào)或提交EMA仲裁,。這種多層級審評機制要求申請人在文件準(zhǔn)備階段即考慮區(qū)域兼容性,,避免后續(xù)流程延誤。 eCTD4.0的探索與未來方向 ICH于2015年發(fā)布的eCTD4.0版本旨在簡化目錄結(jié)構(gòu),、支持多產(chǎn)品類型(如醫(yī)療器械)申報,,并增強生命周期管理功能,。歐盟計劃通過2024年試點逐步過渡至4.0,,其扁平化文件組織方式有望減少重復(fù)提交并提升審評效率。然而,,實施需解決現(xiàn)有系統(tǒng)兼容性及行業(yè)適應(yīng)性問題,。美國eCTD驗證標(biāo)準(zhǔn)相關(guān)技術(shù)支持。河北eCTD注冊系統(tǒng)
中DMF注冊申報相關(guān)技術(shù)支持。工業(yè)園區(qū)中國eCTD便宜
設(shè)施費動態(tài)調(diào)整 API工廠和制劑工廠年費分別約6.8萬和14.5萬美元(2025財年),,CMO工廠費用為制劑費的24%,。國外工廠需額外支付1.5萬美元跨境檢查費,。 ?繳費時限與懲罰 費用需在財年首日(10月1日)起20天內(nèi)繳納,,逾期將列入拖欠名單并暫停ANDA受理,涉事藥品視為冒牌產(chǎn)品,。 ?豁免與特殊情形 PET藥物,、非商業(yè)產(chǎn)品及停產(chǎn)超一年的工廠可豁免繳費,。已繳費工廠若年度內(nèi)無生產(chǎn)活動,仍需繳納費用,。 ?行業(yè)影響與策略 費用上漲推動企業(yè)優(yōu)化申報策略,,例如集中ANDA提交周期、采用CMO外包降低設(shè)施費,,并通過預(yù)認(rèn)證(如DMF完整性評估)減少重復(fù)支出,。 工業(yè)園區(qū)中國eCTD便宜
- 高新區(qū)中國eCTD歡迎選購 2025-05-14
- 靜安區(qū)生物制品eCTD使用 2025-05-14
- 蕪湖新藥eCTD是什么 2025-05-14
- 吳江區(qū)賦悅科技eCTD供應(yīng)商 2025-05-14
- 南京生物制品eCTD注冊系統(tǒng) 2025-05-14
- 上海化學(xué)藥品eCTD格式 2025-05-09
- 南京電子申報eCTD哪個品牌好 2025-05-09
- 太倉NDAeCTD服務(wù)價格 2025-05-09
- 浦東新區(qū)原料藥eCTD文件如何制作 2025-04-26
- 南京新藥eCTD找哪家 2025-04-26
- 品牌企業(yè)管理服務(wù) 2025-06-05
- 徐州怎樣電話交換系統(tǒng)廠家直銷 2025-06-05
- 爆款I(lǐng)P打造的流程 2025-06-05
- 寶山區(qū)品牌科學(xué)計算軟件價格 2025-06-05
- 南京3d虛擬拍攝服務(wù) 2025-06-05
- 上海網(wǎng)絡(luò)機房工程施工 2025-06-05
- 省去硬件SodoCloud技術(shù)白皮書 2025-06-05
- 內(nèi)蒙古國產(chǎn)推廣代理品牌 2025-06-05
- 濟南長清區(qū)B2B網(wǎng)絡(luò)推廣聯(lián)系人 2025-06-05
- 黑河現(xiàn)代化自媒體營銷推廣 2025-06-05