-
全自動金相切割機的切割精度與穩(wěn)定性分析-全自動金相切割機
-
全自動顯微維氏硬度計在電子元器件檢測中的重要作用
-
全自動顯微維氏硬度計:提高材料質(zhì)量評估的關鍵工具
-
全自動維氏硬度計對現(xiàn)代制造業(yè)的影響?-全自動維氏硬度計
-
跨越傳統(tǒng)界限:全自動顯微維氏硬度計在復合材料檢測中的應用探索
-
從原理到實踐:深入了解全自動顯微維氏硬度計的工作原理
-
全自動金相切割機在半導體行業(yè)的應用前景-全自動金相切割機
-
全自動金相切割機的工作原理及優(yōu)勢解析-全自動金相切割機
-
全自動洛氏硬度計在材料科學研究中的應用?-全自動洛氏硬度計
-
全自動維氏硬度計在我國市場的發(fā)展現(xiàn)狀及展望-全自動維氏硬度計
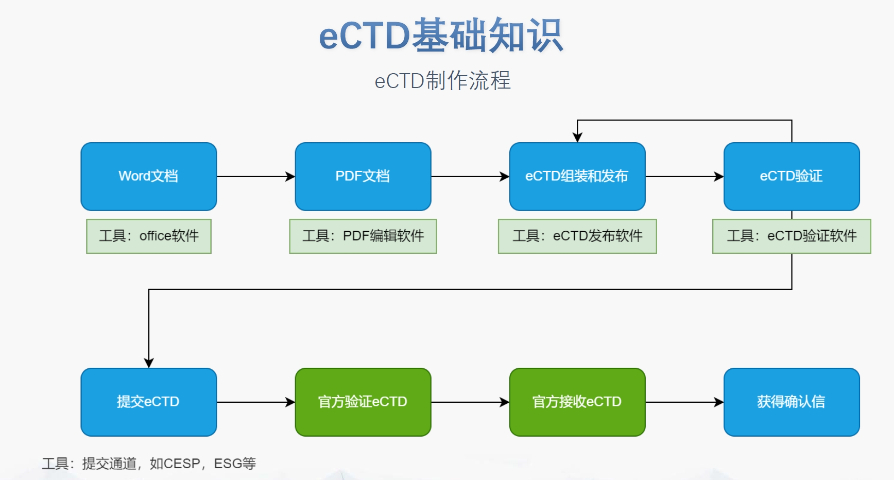
工業(yè)園區(qū)國內(nèi)注冊eCTD業(yè)務
GDUFA III框架與費用分類 2022年更的GDUFA III將費用分為ANDA申請費,、DMF認證費、項目費及設施費四類,,實施周期至2027年,。2025財年ANDA費用漲至約22萬美元,較2024年增幅達27.5%,,反映審評成本上升,。 ?ANDA申請費規(guī)則 費用需在提交時繳納,若申請被拒可退還75%,。重提交視為申請,,需再次繳費。關聯(lián)API的工廠數(shù)量影響總費用,,例如某ANDA引用3個API且涉及6家工廠,,需支付6倍DMF費用。 ?DMF費用機制 II類原料藥DMF需在引用前繳費,,一次性支付約5.3萬美元(2025財年),。未繳費DMF不得用于支持ANDA,否則觸發(fā)退審,。 ?項目費分級管理 根據(jù)企業(yè)獲批ANDA數(shù)量分為大,、中,、小型三級,2025年大型企業(yè)年費約34萬美元,。附屬公司ANDA數(shù)量合并計算,,繳費責任可由母公司或任一附屬公司承擔。加拿大ANDA注冊申報相關技術支持,。工業(yè)園區(qū)國內(nèi)注冊eCTD業(yè)務

美國eCTD的強制實施時間與范圍:美國自2017年5月5日起要求藥申請(NDA),、仿制藥申請(ANDA)和生物制品許可申請(BLA)必須通過eCTD格式提交,2018年5月5日進一步擴展至臨床試驗申請(IND)和藥品主文件(DMF),。FDA通過《聯(lián)邦食品、藥品和化妝品法案》第745A條明確電子提交的強制性,,豁免非商業(yè)化IND和部分DMF類型(如Ⅲ類),。2023年數(shù)據(jù)顯示,F(xiàn)DA接收的eCTD申請占比已達92%,,標志著電子化審評體系的成熟,。企業(yè)若未按規(guī)范提交(如缺少文件或重復序列號),將直接被拒收,。江蘇國產(chǎn)eCTD品牌歐盟IND注冊申報相關技術支持,。
歐盟eCTD的歷史沿革與強制實施 歐盟自2003年逐步推進eCTD(電子通用技術文檔)的標準化進程,初要求藥注冊申請(MAA)采用CTD格式,。2010年,,集中審評程序(CP)率先強制使用eCTD,隨后分散程序(DCP)和互認程序(MRP)分別于2015年,、2017年跟進,。至2019年,歐盟要求所有國家程序(NP)的注冊申請均以eCTD格式提交,,標志著其電子遞交體系的成熟,。2024年,EMA啟動eCTD4.0試點項目,,旨在提升技術兼容性與審評效率,。 eCTD驗證標準的迭代與關鍵更 歐盟的驗證標準歷經(jīng)多次調(diào)整,例如2025年3月啟用的eCTD3.1區(qū)域模板和驗證規(guī)則v8.1,,對文件結構,、元數(shù)據(jù)和內(nèi)容完整性提出更嚴格的要求。標準引入的“追蹤表(Tracking Table)”強制校驗規(guī)則(如15.11和15.12)曾導致CEP(歐洲藥典適用性證書)遞交,,后通過允許占位文件臨時解決,。與早期版本相比,v8.1強化了對模塊一區(qū)域信息的邏輯驗證,,并細化了對PDF書簽,、超鏈接的規(guī)范性檢查,。
經(jīng)濟影響與成本效益 盡管初期投入較高(平均每企業(yè)需50萬歐元),但eCTD可減少30%的審評延遲成本,,長期效益,。仿制藥企業(yè)通過eCTD復用原研數(shù)據(jù),節(jié)省80%的申報準備時間,。歐盟預算撥款2億歐元資助中小企業(yè)完成數(shù)字化轉型,。 倫理審查與數(shù)據(jù)隱私 eCTD中的患者數(shù)據(jù)需匿名化處理,符合《通用數(shù)據(jù)保護條例》(GDPR)要求,。臨床試驗模塊(模塊5)的提交需附帶倫理委員會批準文件,,且區(qū)域版本需體現(xiàn)各國倫理審查差異。AI輔助匿名化工具在保護隱私的同時提升數(shù)據(jù)處理效率,。 技術融合與跨領域應用 eCTD格式擴展至醫(yī)療器械和保健品領域,,歐盟試點eCTD-MDR項目整合ISO標準?;虍a(chǎn)品的eCTD需附加生物安全數(shù)據(jù)庫,,并與歐盟基因庫實時同步。未來,,eCTD或與電子健康檔案(EHR)系統(tǒng)對接,,支持個性化用藥。 持續(xù)改進與行業(yè)反饋機制 EMA每年發(fā)布eCTD實施報告,,分析常見錯誤并更指南,。行業(yè)聯(lián)盟(如EFPIA)通過定期研討會向監(jiān)管機構反饋技術痛點,推動標準優(yōu)化,。開放式API接口的推廣將促進eCTD工具鏈的互操作性,,降低技術鎖定風險。歐盟CESP提交通道相關技術支持,。
美國于2003年成為全球早采用eCTD(電子通用技術文檔)的國家之一,,初由CDER和CBER作為電子提交平臺試點。2008年起,,eCTD正式成為藥申請(NDA)和生物制品許可申請(BLA)的標準格式,,并在2012年通過《藥申報者付費法案》(PDUFA)進一步強化其法律地位。至2017年,,F(xiàn)DA強制要求所有藥申請(NDA),、簡略藥申請(ANDA)及藥物主文件(DMF)必須采用eCTD格式提交,標志著其從可選到強制的轉型,。這一進程在2018年擴展至臨床試驗申請(IND),,終實現(xiàn)全類型藥品注冊的電子化覆蓋澳大利亞eCTD申報軟件相關技術支持。蘇州國際注冊eCTD注冊系統(tǒng)
澳大利亞eCTD注冊外包相關技術支持。工業(yè)園區(qū)國內(nèi)注冊eCTD業(yè)務
2015年發(fā)布《關于藥品醫(yī)療器械審評審批制度的意見》,,提出藥監(jiān)五大目標,,將eCTD納入國家藥監(jiān)數(shù)字化戰(zhàn)略。2017年,,中國加入ICH(國際人用藥品注冊技術協(xié)調(diào)會),,成為全球第八個監(jiān)管機構成員,加速與國際標準接軌,。2018年,,國家藥監(jiān)局(NMPA)完成eCTD文檔管理系統(tǒng)招標,由上海寶信與德國LORENZ合作搭建技術平臺,,標志著技術基礎設施的落地,。 ?規(guī)范制定與試點階段(2019-2023年)? 2019-2020年,CDE(藥品審評中心)發(fā)布《eCTD技術規(guī)范》《驗證標準》等征求意見稿,,并組織兩輪企業(yè)測試,。2021年,NMPA明確化學藥1類,、5.1類及生物制品1類上市申請適用eCTD。2022年實施電子申報(非eCTD格式),,2023年取消紙質(zhì)資料提交,,為eCTD鋪開奠定基礎。 ?實施與擴展階段(2024-2025年)? 2024年3月更電子申報技術要求,,7月啟動網(wǎng)絡傳輸試點,。2025年1月27日,NMPA將eCTD適用范圍擴大至化藥1-5類臨床試驗及上市申請,、生物制品1-3類全流程,,覆蓋藥、仿制藥及生物類似藥,,實現(xiàn)與國際主流申報模式同步,。工業(yè)園區(qū)國內(nèi)注冊eCTD業(yè)務
- 高新區(qū)中國eCTD歡迎選購 2025-05-14
- 靜安區(qū)生物制品eCTD使用 2025-05-14
- 蕪湖新藥eCTD是什么 2025-05-14
- 吳江區(qū)賦悅科技eCTD供應商 2025-05-14
- 南京生物制品eCTD注冊系統(tǒng) 2025-05-14
- 上海化學藥品eCTD格式 2025-05-09
- 南京電子申報eCTD哪個品牌好 2025-05-09
- 太倉NDAeCTD服務價格 2025-05-09
- 浦東新區(qū)原料藥eCTD文件如何制作 2025-04-26
- 南京新藥eCTD找哪家 2025-04-26
- 重慶多功能文件備份-網(wǎng)盤常用知識 2025-06-05
- 靜安區(qū)品牌數(shù)據(jù)定向分析 2025-06-05
- 閔行區(qū)電商平臺代運營圖片 2025-06-05
- 靜安區(qū)怎樣科學計算軟件供應 2025-06-05
- 松江區(qū)互聯(lián)網(wǎng)數(shù)據(jù)服務誠信合作 2025-06-05
- 品牌企業(yè)管理服務 2025-06-05
- 徐州怎樣電話交換系統(tǒng)廠家直銷 2025-06-05
- 虹口區(qū)怎樣Matlab供應 2025-06-05
- 爆款IP打造的流程 2025-06-05
- 寶山區(qū)品牌科學計算軟件價格 2025-06-05