���KNDAeCTD�N���Ԓ ���]��ԃ �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-30

�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-30
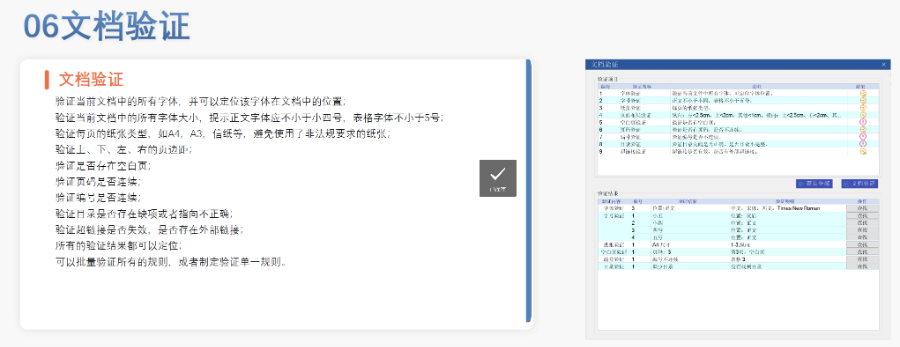
eCTD�ķ�Ҏ(gu��)����c���g(sh��)Ҏ(gu��)�����W��eCTD�ķ�Ҏ(gu��)�Ӽ�����ָ�ϣ�Guidelines��,��ָ�Directive���ͷ�Ҏ(gu��)��Regulation��,�����У���Ҏ(gu��)����CTR������ֱ�ӷ���Ч��,����ָ�ϣ���ICH eCTDҎ(gu��)�����t�鼼�g(sh��)�����ṩ����,��eCTD�ĽY(ji��)��(g��u)����ϚW��ģ�K1Ҏ(gu��)����DTD 3.0+�������������ļ���ģ�K1��,���|(zh��)����(sh��)��(j��)��ģ�K3�����R���о���棨ģ�K5���ȃ�(n��i)��,����ͨ�^XML�ļ����F(xi��n)��(sh��)��(j��)��(li��n)������,��CEP���W��ˎ���m�����C������eCTD�����Ϊ���(g��u)���ŷ⣨Envelope����ģ�K1,����ָ�����R����UUID���Դ_�����g(sh��)��Ҏ(gu��)�ԡ���ʿDMFע��������P(gu��n)���g(sh��)֧��,�����KNDAeCTD�N���Ԓ

�W��eCTD���f��;���c���g(sh��)Ҫ�� ��ͬ���u����?q��)��?y��ng)��ͬ�f�����������г���CP��ͨ�^EMA��eSubmission Gateway��Web Client�ύ,����ɢ����DCP���ͻ��J����MRP���t��ʹ�ÚW��ͨ���ύ�T����CESP�����ļ��Y(ji��)��(g��u)�������ѭģ�K��Ҫ��,������CEP��Ո�����ģ�K1�������ļ���,��ģ�K2���|(zh��)����������ģ�K3�����g(sh��)�ęn������XML�����ļ�횷���EDQM���ض�����Ҏ(gu��)�t,������,������PDF�ļ���o�ܴa���o����ȫ�ęz��,����Ƕ��Ӽ�������֧�ֿ��ٌ��,�� CEP��Ո��eCTD�f�������� CEP������2018�������Ʋ���eCTD��ʽ�����c�u��ԭ��ˎ�Ƿ���ϚW��ˎ��˜�,����ģ�K1�����EDQM��Ո��,�����v��׃���f����,��ģ�K2��ʹ��EDQM�ṩ���|(zh��)������ģ�壬ģ�K3�t��CTD��ʽ�M��3.2.S�¹�(ji��)��(n��i)��,��CEP�cASMF���������|(zh��)���ļ�������Ҫ�^(q��)�e�����ԣ�CEP�o���P(gu��n)(li��n)�����S��,���Ҍ��u��EDQM��ɡ��㽭CDE eCTD�ԃr�ȼ��ô�INDע��������P(gu��n)���g(sh��)֧��,��
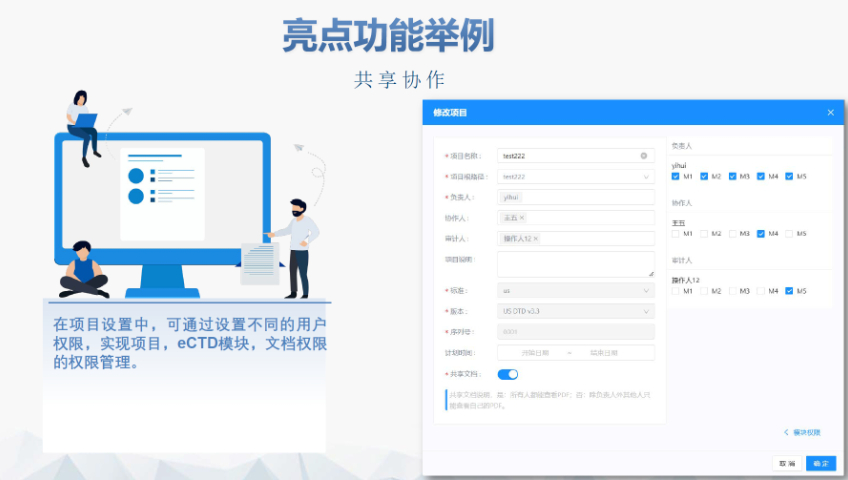
�x��Word��� �����аl(f��)Word��� ���پ�������word���ù��ܰ��o,�������l���ГQ�ˆΣ���(n��i)�Ø��},������,�����֡�Ŀ�,����朽ӵȵĸ�ʽ�͘�ʽ,���ɿ����O(sh��)�ú��ęn�ĸ�ʽ ����朽ӣ��p��������ק�ķ�ʽ�������ı���朽ӻ����}ע��朽�,��������ȫ���P(gu��n)�I��,���Ԅ�������朽� �ęn��֣��ɸ���(j��)��ͬ�ėl����word�ļ��w����,����ֹ�(ji��)��,���ü����_,��퓴a�������Զ��x퓴a�� PDF�D(zhu��n)�Q��WORD�D(zhu��n)PDF,���Ԅ��Д��Ƿ����ɕ������Ԅ��Ƕ�������w,������PDF���پW(w��ng)퓞g�[��PDF,���_�����ɵ�PDF���Ќ��Է��Ϸ�Ҏ(gu��)Ҫ�� �ęn��C����C�ęn�����w����̖,������,����沼�֡��հ��,��퓴a,����̖��Ŀ�,����朽ӵ�,�����ҿ��Զ�λ��C�Y(ji��)�� �ɶ��ƣ��ɸ���(j��)�Ñ������Ƹ�ʽ�͘�ʽģ��
������2003��ɞ�ȫ�������eCTD�����ͨ�ü��g(sh��)�ęn���ć���֮һ������CDER��CBER��������ύƽ�_ԇ�c,��2008����,��eCTD��ʽ�ɞ�ˎ��Ո��NDA����������Ʒ�S����Ո��BLA���Ę˜ʸ�ʽ,������2012��ͨ�^��ˎ����߸��M��������PDUFA���Mһ�������䷨�ɵ�λ,����2017��,��F(xi��n)DA����Ҫ������ˎ��Ո��NDA��������ˎ��Ո��ANDA����ˎ�����ļ���DMF����횲���eCTD��ʽ�ύ,����־����Ŀ��x�����Ƶ��D(zhu��n)��,���@һ�M����2018��Uչ���R��ԇ���Ո��IND�����K���F(xi��n)ȫ���ˎƷע�Ե���ӻ����weCTDע��������P(gu��n)���g(sh��)֧��,��
��(j��ng)��Ӱ��c�ɱ�Ч�� �M�ܳ���Ͷ���^�ߣ�ƽ��ÿ��I(y��)��50�f�WԪ��,����eCTD�ɜp��30%�Č��u���t�ɱ����L��Ч��,������ˎ��I(y��)ͨ�^eCTD��(f��)��ԭ�Д�(sh��)��(j��),����(ji��)ʡ80%�����ʂ�r�g���W���A(y��)��ܿ�2�|�WԪ�Y����С��I(y��)��ɔ�(sh��)�ֻ��D(zhu��n)��,�� ���팏���c��(sh��)��(j��)�[˽ eCTD�еĻ��ߔ�(sh��)��(j��)��������̎��,�����ϡ�ͨ�Ô�(sh��)��(j��)���o�l������GDPR��Ҫ���R��ԇ�?z��i)��K��ģ�K5�����ύ�踽������ί�T�������ļ�,���҅^(q��)��汾���w�F(xi��n)�������팏��,��AI�o�������������ڱ��o�[˽��ͬ�r������(sh��)��(j��)̎��Ч�ʡ� ���g(sh��)�ں��c���I(l��ng)��(y��ng)�� eCTD��ʽ�Uչ���t(y��)����е�ͱ���Ʒ�I(l��ng)��,���W��ԇ�ceCTD-MDR�Ŀ����ISO�˜�,������a(ch��n)Ʒ��eCTD�踽�����ﰲȫ��(sh��)��(j��)��,�����c�W�˻���쌍�rͬ��,��δ����eCTD���c��ӽ����n����EHR��ϵ�y(t��ng)����,��֧�ւ��Ի���ˎ,�� ���m(x��)���M�c�ИI(y��)�����C�� EMAÿ��l(f��)��eCTD��ʩ��棬������Ҋ�e�`����ָ��,���ИI(y��)(li��n)�ˣ���EFPIA��ͨ�^������ӑ����O(ji��n)�ܙC��(g��u)�������g(sh��)ʹ�c,���ƄӘ˜ʃ�(y��u)�����_��ʽAPI�ӿڵ��ƏV�����MeCTD����朵Ļ�������,�����ͼ��g(sh��)�i���L(f��ng)�U,���Ĵ�����NDAע��������P(gu��n)���g(sh��)֧�֡����K����ˎeCTD����(w��)���Ŀɿ�
�Ĵ�����DMFע��������P(gu��n)���g(sh��)֧��,�����KNDAeCTD�N���Ԓ
�x��eCTDϵ�y(t��ng) �ļ���C�c�ޏ�(f��) ֧���Ԅ���C�ļ���ʽ����PDF����,�����wǶ�롢��朽������Եȣ�,����һ�I�ޏ�(f��)�����Ϸ�Ҏ(gu��)Ҫ����ļ�,�����磬ϵ�y(t��ng)���Ԅәz��XML�����ļ��ĽY(ji��)��(g��u)��Ҏ(gu��)�ԣ��_�������Ї�,������,���W�˵ȵ^(q��)��eCTD��Ҏ(gu��)�˜ʡ� eCTD�M�b�c�l(f��)�� ���Ԅ����ɷ���CTD�Y(ji��)��(g��u)������ęn��,������XML�����ļ�,���ļ��A����Ҏ(gu��)��������̖����������Ո?zh��)?����̖�ļ��A�Ԅ����ɣ�����֧�ֳ�朽Ӻ͕�����������(chu��ng)��,������,�������ύ������̖��0000�����m(x��)ÿ���ύ�Ԅ��f��,�� �������ڹ��� ֧���ļ�ȫ�������ڲ�������,�����a����Q,���h����,����ͨ�^����̖�B��ֱ�^�@ʾ���¹�(ji��)�ļ�����Ч�ԣ����w�ij����ύ�����,�����е�ȫ���̹���,�� �f(xi��)ͬ�c��(qu��n)���� ����B/S�ܘ�(g��u)���g�[��/����(w��)������֧���ƶ˻��`���,��ȫ���F�~̖ͨ��,���ṩ���Ñ�f(xi��)�����ܣ�������(qu��n)�ּ�,����Ӌۙ,���ļ��汾���Ƶȡ� ��Ҏ(gu��)֧���c���I(y��)����(w��) ��(n��i)�÷����Ї�CDE,������FDA,���W��EMA�ȷ�Ҏ(gu��)��ģ�壬ͬ�r�ṩע����ԃ,���Y����,��eCTD��ʽ���D(zhu��n)��ȫ����֧�֣��Fꠓ���17��ˎƷע�Խ�(j��ng)�,�����KNDAeCTD�N���Ԓ